





- Wet preparation of calcium phosphates from aqueous solutions
Byeong Woo Lee* and Il Gok Hong
Dept. of Ocean Advanced Materials Convergence Engineering, Korea Maritime and Ocean University, Busan 49112, Korea
Abstract. Calcium phosphates such as HA (hydroxyapatite), β-TCP (tricalcium phosphate) and biphasic HA/β-TCP, were synthesized
by wet chemical precipitation in aqueous solution combined with ball milling
process. Nanosize powders of the calcium phosphates were synthesized using
Ca(OH)2 and H3PO4. The effects of initial precursor Ca/P ratio (1.30, 1.50 and 1.67),
ball milling process and post heat-treatment on the phase evolution behavior of
the powders were investigated. The phase of resulting powder was controllable
by adjusting the initial Ca/P ratio. HA was the only phase for as-prepared
powders in both cases of Ca/P ratios of 1.50 and 1.67. The single HA phase
without any noticeable second phase was obtained for the initial Ca/P ratio of
1.67 in the overall heat-treatment range. Pure β-TCP and biphasic calcium phosphate (HA/β-TCP) were synthesized from precursor solutions having Ca/P molar ratios of 1.30 and 1.50, respectively, after having been heat-treated above 700 oC. The β-TCP phase has appeared on the pre-existing DCPD (dicalcium
phosphate dihydrate) and/or HA phase. Dense ceramics having translucency were
obtained at a considerably lower sintering temperature. The modified process
offered a fast, convenient and economical route for the synthesis of calcium phosphates.
Keywords: Calcium phosphate, Hydroxyapatite, Tricalcium phosphate, Nanopowder, Wet chemical synthesis
Calcium phosphates have
been widely utilized in biological applications. Among the
calcium phosphates, significant attention has been given to
hydroxyapatite (HA, Ca10(PO4)6(OH)2) and tricalcium phosphate (β-TCP,
Ca3(PO4)2) due to their
prominent bioactivity and biocompatibility [1-4]. Over the past few decades, the biphasic calcium phosphate
(BCP) composed of both the tricalcium phosphate (β-TCP)
and hydroxyapatite (HA) has also received considerable attention due to its
enhanced biocompatibility and mechanical properties [5-7].
Recently, nanometer-sized calcium
phosphates have received much attention due to
their significantly enhanced
efficacy [1, 6]. Solution processes have been widely used
to prepare nanometer-sized powders.
Wet chemical precipitation using aqueous solutions
is the most common synthesis route for the preparation of calcium phosphates nanoparticles, which provides additional advantageous
features such as simple, low cost and
low temperature synthesis [8-10].
Calcium hydroxide (Ca(OH)2) and phosphoric acid
(H3PO4) were widely used as
starting materials for the precipitation process for HA
and β-TCP preparation according to the reactions, Eq. (1) and (2), respectively. The only byproduct of the reactions is water, and the reactions
involve no foreign elements.
10 Ca(OH)2 + 6 H3PO4 → Ca10(PO4)6(OH)2
(HA) + 18 H2O (1)
3 Ca(OH)2+ 2 H3PO4 → Ca3(PO4)2
(TCP) + 6 H2O (2)
The properties of particles prepared by the wet synthesis
method are largely dependent on
process parameters. The phase, size, shape, and surface area of the particles obtained by
the wet process are highly sensitive to the
precursor concentration, Ca/P ratio
of precursors, and reaction pH [5, 11]. Furthermore, the phosphoric acid addition rate is strongly linked
to the suspension stabilization, and also to the final precipitate (powder) properties
[12, 13]. The precipitation chemistry for the preparation of calcium
phosphates has been found to be complicated due to the slow attainment of equilibrium and the amorphous nature of the
products formed. It is also very important to obtain a large amount of products at a
constant reactant volume.
The present study
deals with the modified synthesis of
calcium phosphate powders using a wet precipitation method combined with mechanical ball milling process. In this study, calcium hydroxide was used as a calcium
precursor and phosphoric acid was added as a source of phosphorus according to Eq.
(1) and (2). This work is concerned with a more convenient approach to the
preparation of calcium phosphate compounds such as pure β-TCP, biphasic calcium phosphate (HA/β-TCP) and pure HA, from precursor Ca/P molar
ratios of 1.30, 1.50
and 1.67, respectively, using precursor solutions with
higher concentrations than other wet preparation processes. In order to clarify the effect of precursor Ca/P ratio on the powder
properties, this study has been carried out without any capping or pH control
agents. Nanoscale calcium phosphate powders
were synthesized via the
precipitation process combined with a ball milling process, and the feasibility
of the process for large-scale production at high production rates was
discussed.
Calcium phosphate nanoparticles were prepared by a wet
precipitation route followed by ball milling process under
controlled synthesis conditions. Calcium hydroxide (Ca(OH)2) and
phosphoric acid (H3PO4) were used as precursors.
In the process, 85% of H3PO4 solution was added dropwise
into 275 ml of a high concentration (1 M) of Ca(OH)2 aqueous suspension solutions
within 20 min. For the preparation of the different calcium
phosphates, the predetermined Ca/P molar ratio of the
solutions of 1.30, 1.50 and 1.67 was set by adding
the H3PO4
solution to the continuously stirred Ca(OH)2 suspension at room temperature. The precipitation products with the mother solution were ball milled at 250 revolutions per minute. After the ball milling process up to 12 h, the synthesized powders were dried at 60 oC for 12 h. Unlike other preparation processes for calcium
phosphates, there was no maturing
(aging) and filtering (washing) process involved.
The prepared powders were calcined at different
temperatures ranging from 500 oC to 900 oC to study phase
developments. The dried powders were
formed to pellets, and then sintered at 1,100 oC for 2 h. The properties for the powders
and sintered ceramics were characterized
using scanning electron microscopy (SEM) and X-ray diffraction (XRD) using Cu Kα
radiation (λ = 1.5418 Å). The
particle sizes of the synthesized powders were also
calculated by the XRD peak broadening according to the Scherrer’s equation (Dcal
(particle size) = 0.89 λ/(B cos θ):
where λ is the wavelength, B is the full width at half maxima (FWHM) in radians
and θ is the
diffraction angle).
In the present study, calcium
phosphates such as β-TCP, biphasic
calcium phosphate (HA/β-TCP) and HA were prepared by a
new approach of the aqueous precipitation method, which was combined with mechanical ball milling process, from precursor Ca/P
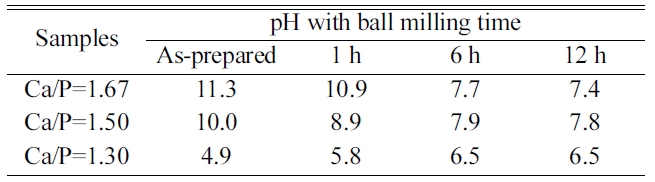
ratios of 1.30, 1.50 and 1.67, respectively. As the Ca/P ratio decreased from 1.67 to 1.30, the
pH after the precipitation reaction decreased from 11.3 to 4.9, due to the lowered base/acid ratios in the solutions. Changes in the pH value of
the solutions as-precipitated and after the ball
milling process are given in Table 1. The initial pH
value of the solution after precipitation was
measured to be 11.3 and the value decreased
to 7.4 after the ball milling process for Ca/P
molar ratio of 1.67. The pH values of biphasic HA/β-TCP also decreased from 10.0 to 7.8 at Ca/P molar ratio 1.5. This decrease in pH values was due to the completion of the chemical reactions of the unreacted precursors through the ball milling process forming HA (i.e. calcium
phosphate-hydroxide) with consumption
of free hydroxide (OH-) in the reactant solution in accordance with Eq. (1). In contrast, for β-TCP
powders from Ca/P molar ratio of 1.30, the pH value was
increased from 4.9 to 6.5 with increasing ball milling time,
because, after the precipitation reaction progressed,
water having the higher pH value (~7) was formed from the lower pH precursor
solution, following Eq. (2).
The effect of ball milling on the phase pure HA
preparation is presented in Fig. 1. When the powder
was prepared in a short period of precursor mixing time within 10 min from Ca/P
of 1.67, the predominant phase of the precipitate was mostly undesirable DCPD (CaHPO4·2H2O (JCPDS 11-293)). Once the DCPD appeared, the phase was quite stable.
To turn the phase into the desired HA phase (hexagonal HA (JCPDS 9-432)), a
heat-treatment up to 1,100 oC
was required as shown in Fig. 1(a). Even after the high temperature heat
treatment, it was difficult to obtain the phase pure HA. On the contrary, the
phase pure HA phase can be synthesized by the simple ball milling process. As
can be seen in Fig. 1(b), there was a drastic phase change from DCPD to HA by
ball milling process. To prepare the single HA phase
precipitate, about 6 hours of ball milling
was required. On the other hand, the β-TCP phase (JCPDS 9-169) could not be
easily synthesized by ball milling process using the precipitates from the
precursor Ca/P ratios of 1.30 and
1.50, and subsequent heat treatment was necessary to prepare biphasic HA/β-TCP
and phase pure β-TCP.
Fig. 2 shows the
XRD patterns of the
calcium phosphate powders after ball milling for 12 h, from the precursor Ca/P ratios of
1.30, 1.50 and 1.67. A mixed phase of DCPD with HA phase was obtained with Ca/P ratio of 1.30.
For the precursor Ca/P ratios of 1.50 and 1.67, the patterns are
showing only the HA phase formed without other
phases. There is no noticeable difference between the peaks for the precipitates prepared. In addition, the broad and weak intensity of the HA peaks indicates poor crystallinity and/or nanometer scale
crystallites of the
calcium phosphate powders prepared. The
prepared powders are approximately 26 nm in particulate size
for both cases calculated by the Scherrer’s
equation. The most distinct (002) reflections (near 2θ 26o) in the
XRD patterns were taken into calculation.
Calcination temperature
plays an important role in the phase formation, as shown
in Fig. 3. In the case of Ca/P =1.30
powder, it showed that phase pure β-TCP was formed by
consuming the pre-existing DCPD and HA as
the calcination temperature increased from 500 oC to 900 oC (Fig. 3(a)). Biphasic
calcium phosphate (β-TCP/HA) and single phase HA can
be obtained from the powders with Ca/P ratios of 1.50 and 1.67, respectively.
Fig. 3(b) shows that in the case of the
powder from Ca/P = 1.50 the TCP phase appears with the pre-existing HA phase after
calcination at 700 oC due to the phase transformation
from amorphous to crystalline β-TCP. The β-TCP
was the dominant phase thereafter. The percentage of volume fraction for β-TCP and HA was
calculated using the relative intensity ratio (RIR) [14]. The HA/β-TCP ratio
was estimated to be 35/65 at 900 oC. For powders from the precursor Ca/P ratio of
1.67, the overall peaks of HA become more distinct with the increasing calcination temperature, and the
narrow peak width suggests increased crystallinity and particulate size (Fig. 3(c)). The particulate size increased from 30 to 50 nm when the calcination
temperature increased from 500 to 900 oC,
respectively. No impurity or secondary phases appeared in this case.
The sinterability of the synthesized powders is
proportional to particle size. The ultra-fine particle size of as-prepared
precipitates leads to higher sinterability. In this study, the diameter of the
green pellets was 12.71 mm. After sintering at 1,100 oC for 2 h, the
diameter has decreased to 9.85 mm (22.5% reduction for HA
from Ca/P=1.67), 9.5 mm (25.3% reduction for β-TCP/HA from Ca/P=1.50) and 10.95
mm (18.5% reduction for β-TCP from Ca/P=1.30). The sintered bodies showed a
certain degree of translucency showing good sinterability (Fig. 4). For pure
HA, it is known that the sintering temperature to achieve such dense ceramics
is 1,250 oC or higher [15, 16]. In particular, low temperature sintering is
essential for β-TCP, because β-TCP reconstructively
transforms into α-TCP above 1,125 oC [17-19].
Ultrafine particles with high reactivity offered high-density ceramic products
at the lower sintering temperature of 1,100 oC. Fig.
5 shows the microstructures of sintered specimens with the precursor
Ca/P ratios of 1.30, 1.50 and 1.67 at 1,100 oC for 2 h. Grains
of around 0.5-1.8 μm in sizes
are observed in the specimens.
In most chemical precipitation processes for calcium
phosphates, the powder properties depend heavily on the processing parameters during
the precipitation reaction due to the slow reaction rate
and the unstable nature of the precursor solution. The morphology, phase, and crystallinity of precipitated
powders were closely
related to precursor concentration,
precursor dropping rate, reaction
pH, and exposure (aging) time in solutions. For example, to prepare single HA phase particles, very slow precursor
dropping rates such as a few drops per minute and a very long aging time over
24 h were essential at low aqueous precursor concentrations of less than 0.6 M
Ca(OH)2 [20, 21].
Furthermore, some wet chemical methods require an extra time-consuming washing step to rinse off by-products and excess reactants. However, in this study, nanometer scale HA, β-TCP, and biphasic β-TCP/HA
powders were conveniently prepared in a fast and simple manner: about 100 times
faster precursor dropping rate at higher precursor concentration without
washing step. Therefore, it can be proposed that the modified process is
economically feasible and capable of mass
production in large quantities.
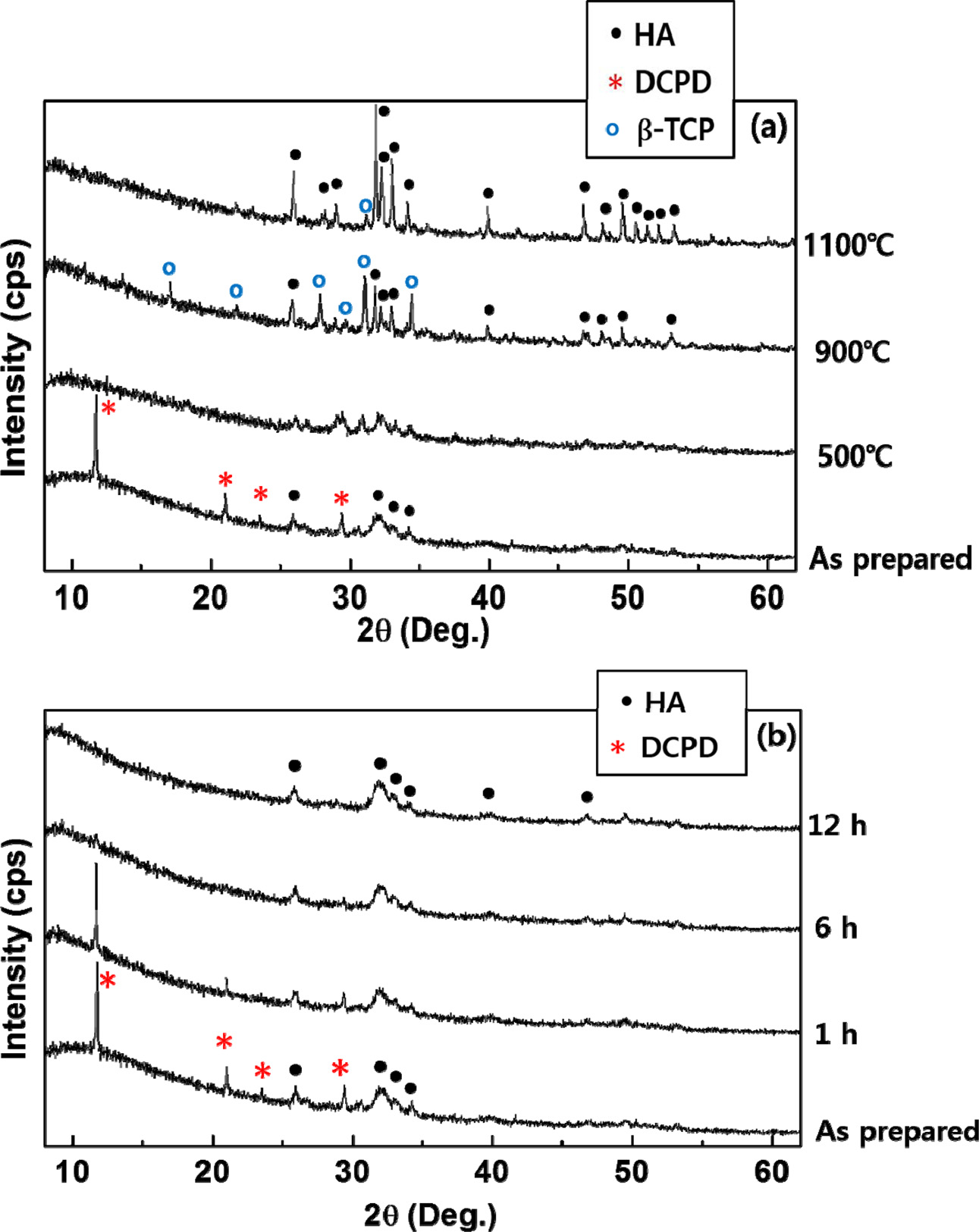
|
Fig. 1 XRD patterns of synthesized HA powders from Ca/P = 1.67 prepared by (a) heat treatment at different temperatures and (b) ball milling process with various milling time. |
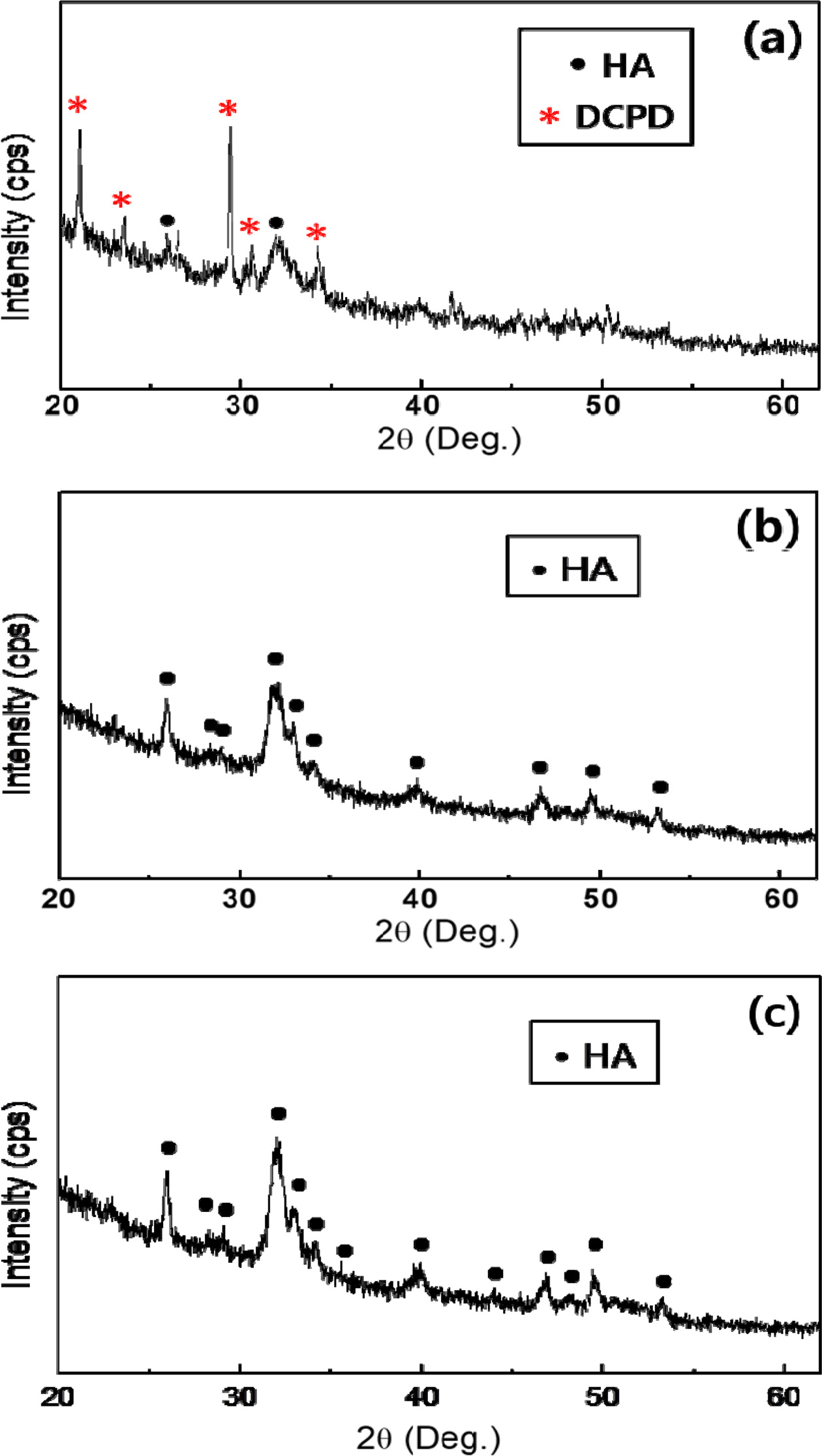
|
Fig. 2 XRD patterns of precipitated calcium phosphates powders from the different precursor Ca/P ratios: (a) Ca/P = 1.30, (b) Ca/P = 1.50, and (c) Ca/P = 1.67. |
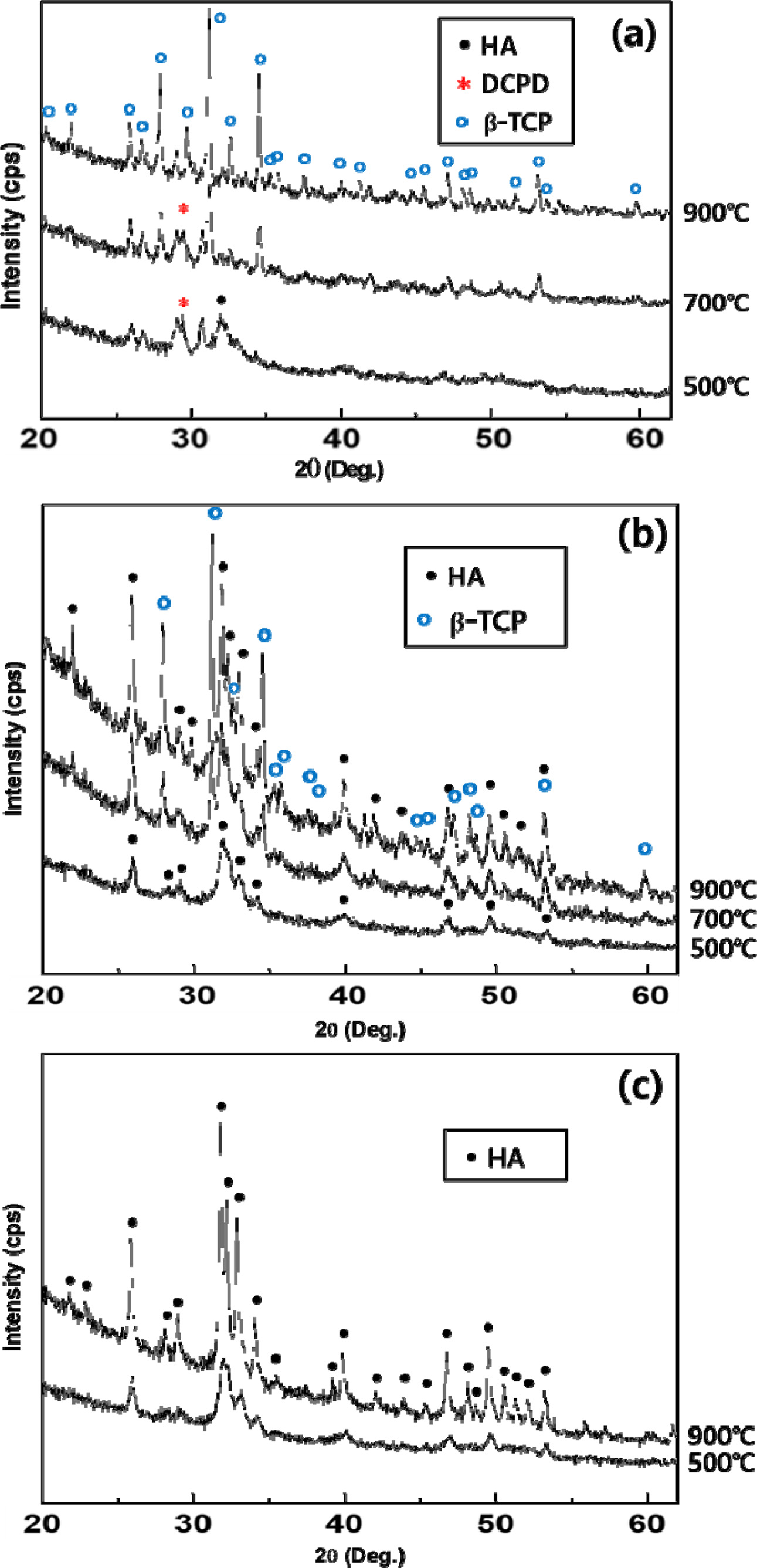
|
Fig. 3 XRD patterns of prepared calcium phosphates powders from the different precursor Ca/P ratios ((a) Ca/P = 1.30, (b) Ca/P = 1.50, and (c) Ca/P = 1.67) calcined at different temperatures. |

|
Fig. 4 A translucency example for the sintered specimens at 1,100 oC for 2 h using the prepared calcium phosphates powders from the different precursor Ca/P ratios (TCP (Ca/P = 1.30), TCP/HA (Ca/P = 1.50), and HA (Ca/P = 1.67)). |

|
Fig. 5 SEM micrographs of the sintered ceramics at 1,100 oC for 2 h using the prepared powders from the different precursor Ca/P ratios: (a) TCP (Ca/P = 1.30), (b) β-TCP/HA (Ca/P = 1.50), and (c) HA (Ca/P = 1.67). |
|
Table 1 pH values with ball milling time of precipitated calcium phosphates powders with different precursor Ca/P ratios: Ca/P = 1.30, 1.50, and 1.67. |
The modified approach of the aqueous precipitation method followed by the mechanical ball milling process offered a simple, convenient and economical
route to synthesize calcium phosphates without the pH
control and ripening time used in most wet precipitation
studies. The ball milling step enhanced the reaction process and
promoted the desirable phase synthesis in a short period of time. The biphasic HA/β-TCP composite and pure HA and β-TCP
powders were synthesized according to the initial Ca/P ratio and heat treatment. It was shown that, as the heat treatment temperature increased, the phase pure β-TCP was formed by consuming the pre-existing DCPD and HA in the case of Ca/P = 1.30 powder. For Ca/P=1.50 powder, the biphasic HA/β-TCP
was obtained, where the TCP phase appeared on the pre-existing HA phase after heat treatment at 700 oC. Pure
HA phase without any noticeable second phase
can be synthesized without post heat treatment for the
precursor Ca/P ratio of 1.67. Due to the high
reactivity of the nanoparticles prepared via the modified route, highly dense
ceramics with translucency were formed at a relatively low sintering
temperature of 1,100 oC.
This work was supported by the National Research
Foundation of Korea (NRF) grant funded by the Korean government (Ministry of
Science and ICT) (No. 2017M2B2A9073125).
- 1. N. Eliaz and N. Metoki, Mater. 10 (2017) 334-407.
-

- 2. W. Habraken, P. Habibovic, M. Epple, and M. Bohner, Mater. Today 19 (2016) 69-87.
-
- 3. A. Dudek and L. Adamczyk, Optica Aplicata. 13 (2013) 143-151.
-
- 4. C. Wu and Y. Xiao, Bone Tissue Regen. Insights 2 (2009) 25-29.
-
- 5. J. Marchi, P. Greil, J.C. Bressiani, A. Bressiani, and F. Muller, Appl. Ceram. Technol. 6 (2009) 60-71.
-
- 6. S. Kannan and J.M.F. Ferreira, Chem. Mater. 18 (2006) 198-203.
-
- 7. E.C. Victoria and F.D. Gnanam, Trends Biomater. Artif. Organs 16 (2002) 12-14.
- 8. J. Marchi, P. Greil, J.C. Bressiani, A. Bressiani, and F. Muller, Int. J. Appl. Ceram. Technol. 6 (2009) 60-71.
-
- 9. L. Sun, L.C. Chow, S.A. Frukhtbeyn, and J.E. Bonevich, J. Res. Natl. Inst. Stand. Technol. 115 (2010) 243-255.
-
- 10. K. Salma, L. Berzina-Cimdina, and N. Borodajenko, Process. Appl. Ceram. 4 (2010) 45-51.
-
- 11. O. Mekmene, S. Quillard, T. Rouillon, J.M. Bouler, M. Piot, and F. Gaucheron, Dairy Sci. Technol. 89 (2009) 301-316.
-
- 12. A.K. Nayak, Inter. J. Chem. Tech. Res. 2 (2010) 903-907.
- 13. D.S. Gouveia, A.H.A. Bressiani, and J.C. Bressiani, Mater. Sci. Forum 530-531(2006) 593-598.
-
- 14. M. Ebrahimi and M. Botelho, Data in Brief 10 (2017) 93-97.
-
- 15. S. Ramesh, K.L. Aw, R. Tolouei, M. Amiriyan, C.Y. Tan, M. Hamdi, J. Purbolaksono, A. Hassan, and W.D. Teng, Ceram. Int. 39 (2013) 111-119.
-
- 16. S. Ramesh, C.J. Gan, L.T. Bang, A. Niakan, C.Y. Tan, J. Purbolaksono, H. Chandran, S. Ramesh, B.K. Yap, and W.D. Teng, J. Ceram. Proc. Res. 16 (2015) 686-689.
- 17. J.H. Welch and J.H. Gutt, J. Chem. Soc. 29 (1961) 4442–4444.
-
- 18. D.M.B. Wolff, E.G. Ramalho1, and W. Acchar, Mater, Sci. Forum 530-531 (2006) 581-586.
-
- 19. R.G. Carrodeguas and S.De Aza, Acta Biomater. 7 (2011) 3536-3546.
-
- 20. M. Sokolova, I. Kreicbergs, V. Zalite, and L. Berzina-Cimdinaa, Eurasia Conference on Chemical Sciences Proc. (2012) 1-1.
- 21. L.T. Bang and R. Othman, Ceram. Silikaty 58 (2014) 157-164.
 This Article
This Article
-
2019; 20(6): 655-659
Published on Dec 31, 2019
- 10.36410/jcpr.2019.20.6.655
- Received on Jun 7, 2019
- Revised on Sep 16, 2019
- Accepted on Sep 26, 2019
Services
- Abstract
introduction
experimental
results and discussion
conclusions
- Acknowledgements
- References
- Full Text PDF
Shared
Correspondence to
- Byeong Woo Lee
-
Dept. of Ocean Advanced Materials Convergence Engineering, Korea Maritime and Ocean University, Busan 49112, Korea
Tel : +82-51-410-4356 Fax: +82-51-404-3986 - E-mail: bwlee@kmou.ac.kr
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr