





- Electrochemical synthesis for a greener future: Insights from Kolbe electrolysis
Abhishek Saxena, Amith Abraham and Byoung-In Sang*
Department of Chemical Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 04763, Republic of Korea
This article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Electrochemical synthesis has gained renewed interest due to advancements in material science and sustainable chemistry. This review explores Kolbe electrolysis, a green and efficient method for decarboxylating carboxylic acids to produce alkanes, as a sustainable renewable, and greener alternative to traditional chemical processes. It examines the mechanistic insights of the process, including reaction pathways, intermediate species, and kinetics. Factors influencing reaction kinetics include concentration, temperature, pressure, electrode materials, and power sources. The review also discusses advancements in electrode materials, such as platinum (Pt) based modified electrodes, boron-doped diamond (BDD), ruthenium oxide (RuO2), coated electrodes, etc., and discusses the significance of green electrolyte (aqueous), as well as innovative electrolyte (ionic liquids, and deep eutectic solvents). Modern electrolysis cell configurations, such as microreactors and continuous flow cells, are examined for their scalability, control over reaction parameters, and reduced side reactions. The review also discusses future challenges and opportunities in leveraging Kolbe electrolysis to electrification of the chemical industry.
Keywords: Electrochemical synthesis, Kolbe electrolysis, Green chemistry, Hydrocarbons, Electrolysis cell.
Electrochemical methods are crucial in organic synthesis due to their alignment with green chemistry principles, such as waste avoidance, hazardous chemical replacement, safer solvents, energy efficiency, and the use of renewable feedstocks [1, 2]. Electrosynthesis is a sustainable approach that offers precise control over reaction conditions, enhancing selectivity, yield, and efficiency [2, 3]. It can be applied to a variety of organic transformations, making it valuable for synthesizing fine chemicals [4], pharmaceuticals [5], and complex natural products [6]. Industrial applications include metal plating [7], energy storage and conversion [8], waste treatment [9], and electro-organic synthesis.
Electrolysis is a technique used in organic synthesis to convert raw materials into reactive intermediates, achieving high atom economies. It can be accelerated using direct or indirect strategies. Indirect electrolysis involves metal catalysts and organic mediators while direct electrolysis occurs on electrode surfaces without mediators [10]. Kolbe electrolysis has emerged as a clean tool for various applications, including the synthesis of ligands [11], fatty acids [12], benzathine derivatives [13], fuel production, and fine chemicals [14] as well as dimerized silyl acetic acids [15]. The history of electrochemical methods dates back to the 18th century, with key discoveries that laid the groundwork for modern electrochemistry. Luigi Galvani's experiments with "animal electricity" in 1789 sparked an interest in bioelectricity [16]. Alessandro Volta invented the Voltaic Pile in 1800, demonstrating the chemical generation of electricity [17]. Michael Faraday's work in the 1830s established the laws of electrolysis, quantifying the relationship between electric current and chemical changes [18]. Faraday's laws were foundational for understanding the correlation between electrical energy input and chemical changes during electrolysis, critical for chemists like Hermann Kolbe, who explored electrochemical reactions.
Kolbe electrolysis, first described by Hermann Kolbe in 1849, is an electro-organic process that converts carboxylic acids through anodic oxidation [19, 20]. It works with various substrates but requires large current densities and can cause undesired side reactions [21, 22]. A "non-Kolbe" pathway may result from a second oxidation producing a cation, leading to nucleophile addition, rearrangement, or fragmentation [23]. A pseudo-Kolbe reaction occurs when electron transfer from a tethered moiety with a lower oxidation potential occurs. Factors influencing efficiency and selectivity include electrolyte concentration, current density, solution pH, temperature, and pressure [24]. Kolbe electrolysis is a crucial method for symmetrical hydrocarbon synthesis, using electricity as a power source and water as a solvent. It produces various hydrocarbons like alkanes, alkenes, and aromatic compounds while electrolyzing different functional groups like alcohols or ketones. This method minimizes waste and uses milder, more sustainable reagents compared to traditional synthesis routes [25-27]. Unlike electrochemical reduction, which requires specific catalysts and controlled conditions [28], Kolbe electrolysis uses readily available carboxylate salts under milder conditions. It operates at lower temperatures, minimizing side reactions and enhancing energy efficiency [21, 29]. This review article emphasizes recent developments in Kolbe electrolysis, providing a mechanistic overview and discussing its impact on reaction kinetics, advancement in electrode materials as well as electrochemical microreactor technology for industrial scale-up. It concludes with the prospects for the electrification of the chemical industry in the context of Kolbe electrolysis.
Principle of Kolbe electrolysis and formation of product (alkane dimer)
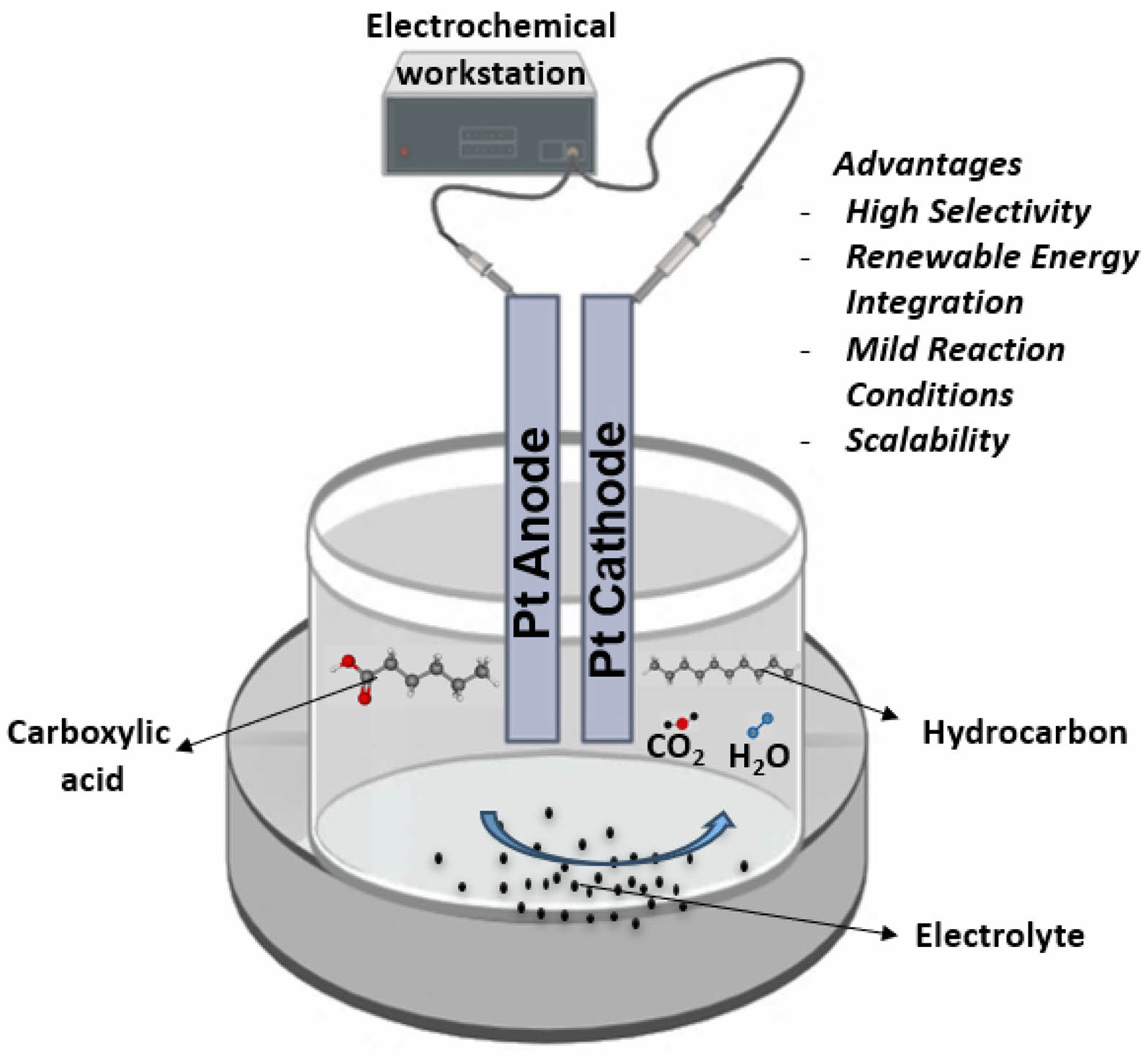
Kolbe electrolysis, named after German chemist Herrmann Kolbe, is an electrochemical conversion of carboxylic acids to valuable products [21]. The anodic oxidation of carboxylic acid derivatives leads to decarboxylation, resulting in key radical intermediates. The process can result in dimerization, unsymmetric radicals, and cyclization processes [10]. It is a crucial reaction in electroorganic synthesis, producing functionalized homo- and hetero-dimers with high selectivity and chirality [30, 31]. During Kolbe electrolysis, the oxygen evolution reaction (OER) is the main side reaction at the anode in aqueous conditions and is suppressed in anhydrous conditions [32-33]. Platinum (Pt) and other noble-metal oxides such as iridium oxide (IrO2), ruthenium oxide (RuO2), and graphene are the most investigated anodes [34]. An electric current causes carboxylic acid to break down into hydrogen, carbon dioxide (CO₂), and an oleaginous liquid [21]. Biomass resources can be used as feedstock in a sustainable and eco-friendly process, addressing the twelve principles of green chemistry and integrating easily into biorefinery concepts for future applications, making it a flexible and environment friendly approach [21, 35]. Fig. 1 presents a schematic overview of the Kolbe electrolysis setup.
The reaction mechanism involves anodic oxidation i.e. deprotonation of carboxylic acid (RCOOH), where carboxylate ions lose electrons to form carboxyl radicals (RCOO•). These radicals undergo decarboxylation, releasing CO₂ and forming alkyl radicals (R•). The alkyl radicals couple to form a new carbon-carbon bond, forming a hydrocarbon (R-R) dimer, and hydrogen evolution reaction (HER) occurs at the cathode. The reaction pathway involves several key steps, including:
Anodic oxidation
Formation of carboxylate ions:
The carboxylic acid (RCOOH) dissociates in the electrolyte to form carboxylate ions (RCOO-) and protons (H+).
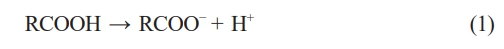
Formation of radical:
The carboxylate ion at the anode surface loses an electron to form a carboxyl radical (RCOO•).

Decarboxylation of the carboxyl radical:
The carboxyl radical quickly loses a CO2 molecule, forming an alkyl radical (R•).

Coupling of alkyl radicals:
The alkyl radicals couple to form a new alkane (R-R).

Cathodic Reduction
At the cathode, water is typically reduced to hydrogen gas and hydroxide ions.
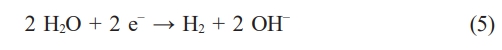
Overall reaction
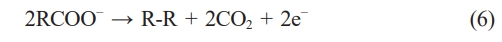
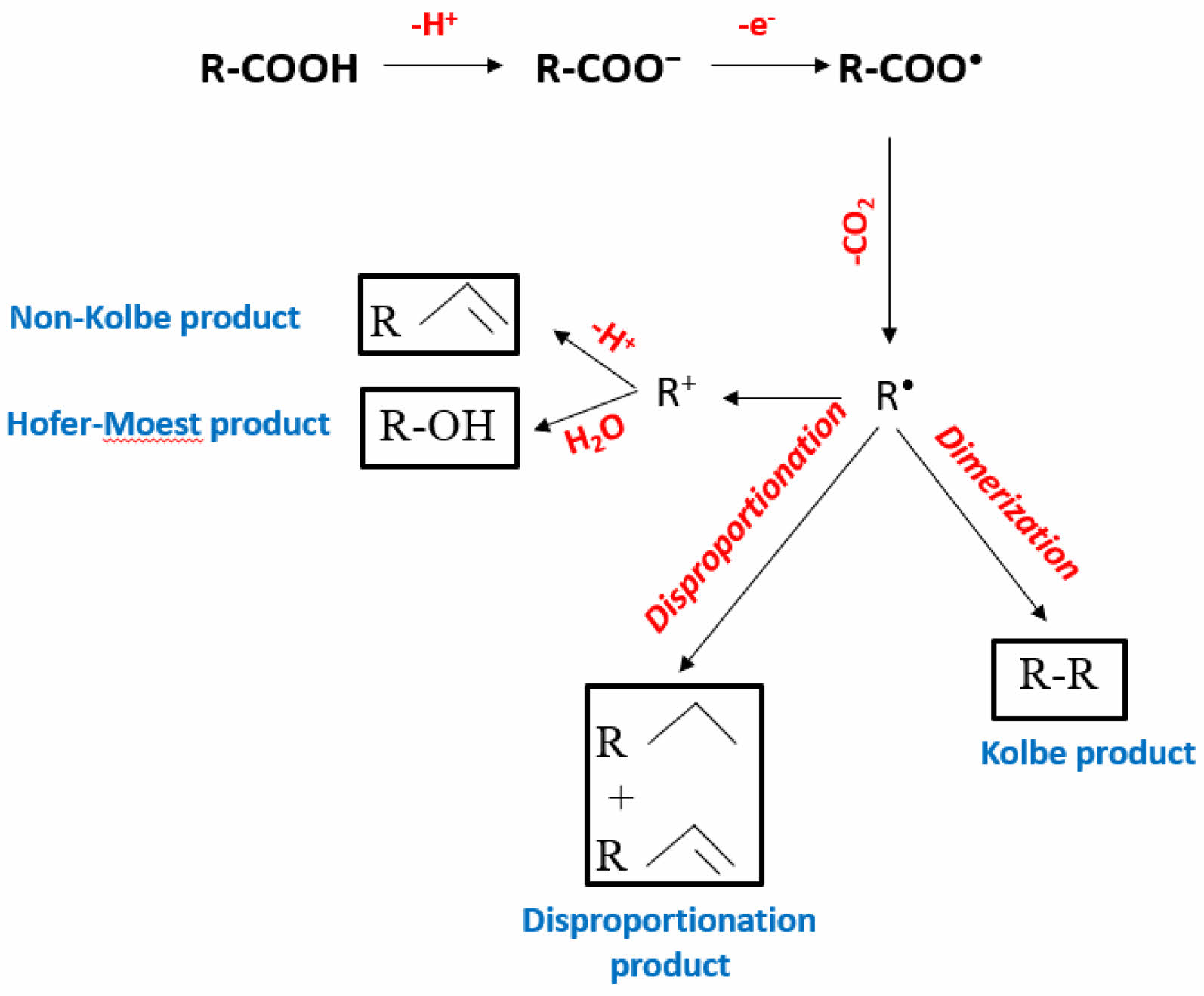
Non-Kolbe product and side reactions
Kolbe electrolysis primarily produces alkane dimers through radical coupling; however, additional oxidation or side reactions, such as the Hofer-Moest reaction, can lead to the formation of non-Kolbe products like esters, alcohols, and alkenes [34, 35]. In these reactions, carboxylate ions undergo decarboxylation, with the resulting radicals either coupling (Kolbe reaction) or undergoing further oxidation to form carbocations R+, leading to non-Kolbe products. Non-Kolbe reactions are particularly effective for anodic oxidation of compounds with α-heteroatoms like lactams and amides, without requiring external catalysts or oxidants [10]. High current densities are crucial for both processes, Pt is commonly preferred for both reactions due to its catalytic efficiency, but carbon electrodes can be used as a cost-effective alternative in non-Kolbe pathways [36]. The schematic reaction mechanism of non-Kolbe electrolysis is shown in Fig. 2.
Factors affecting reaction kinetics (Effect of concentration, Temperature, Pressure, electrode material, and power source)
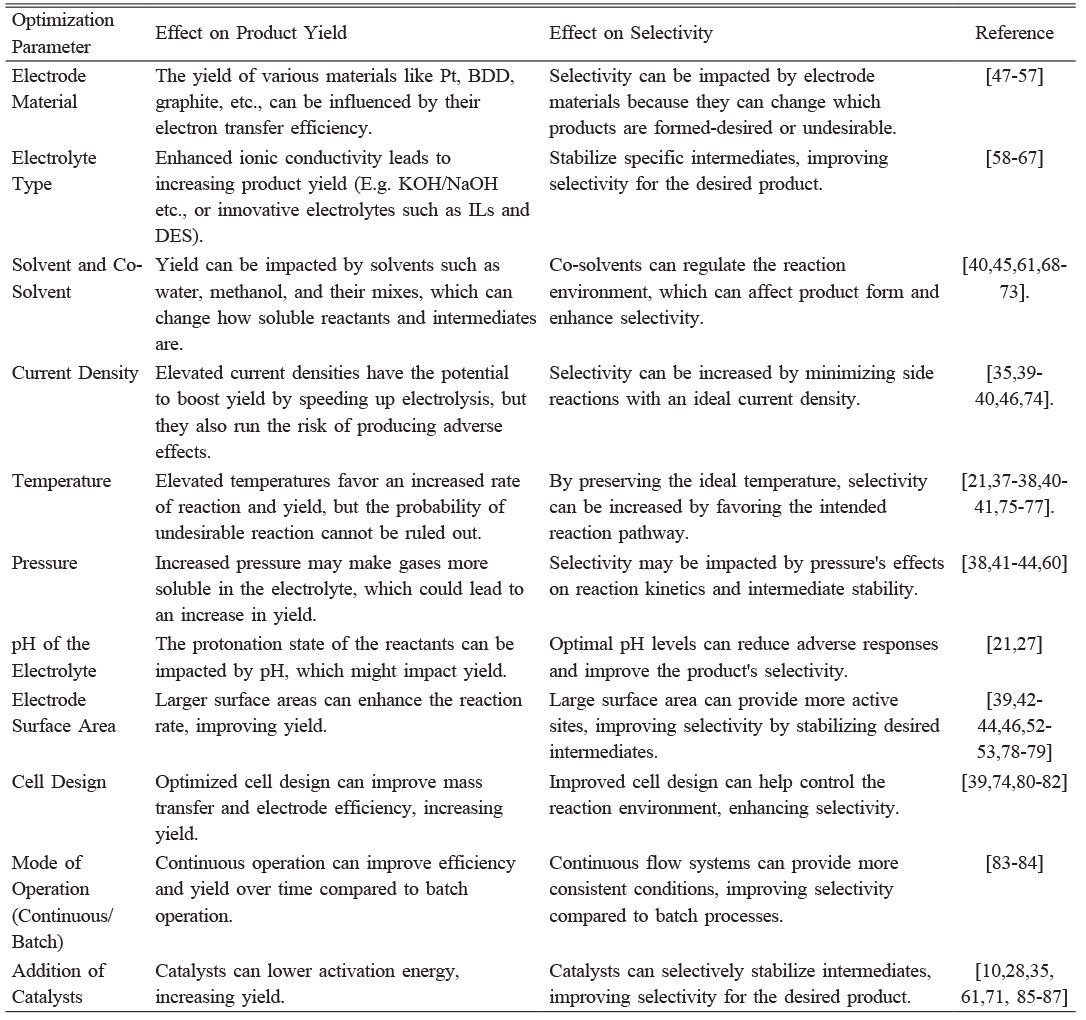
The Kolbe electrolysis reaction's efficiency, selectivity, and success are significantly influenced by reaction conditions such as reactant concentration [37], temperature [21], pressure [38], choice of electrode material [32], and applied current or voltage [39]. These conditions must be optimized to maximize hydrocarbon product yield and minimize side reactions and waste. The Kolbe coupling reaction in an aqueous solution requires high anode current density, a smooth Pt anode, a temperature below 27 °C, a neutral or slightly alkaline medium, and a high carboxylate concentration [21]. Oxygen evolution is the primary reaction at anode potentials up to 2.0 V, with the reaction triggered at higher potentials (2.1 to 3.0 V) and suppressed at lower potentials [40].
Organic acid concentration in Kolbe electrolysis significantly impacts conversion rates, with higher concentrations increasing reactant availability and yield, while excessive concentrations can favor non-Kolbe reactions [37]. Higher temperatures increase reaction rate and produce radicals quickly, but can affect intermediate radical stability, leading to side reactions and reduced hydrocarbon yield. Temperature-dependent selectivity favors different products due to increased energy availability, and temperatures above 50 °C should be avoided to maintain selectivity and product yield [38, 41]. Pressure affects gas solubility in electrolytes, with higher pressures causing more gas dissolution and potentially affecting reaction dynamics and separation efficiency. Lower pressures may cause gas bubble formation, hindering electrode surface area and reaction efficiency. Higher pressures require robust equipment and energy [42-44]. The choice of electrode material significantly impacts electrode kinetics, reaction yield, electrochemical behaviors, current density, and faradaic efficiency, ensuring successful Kolbe product outcomes [32]. The power supply significantly influences the successful outcome of Kolbe electrolysis. The potentiostatic mode uses a potentiostat to apply a constant potential to the electrolyte over time, requiring substrate redox potential knowledge. This mode is more complex and expensive, especially in large-scale cells. Cell current drops when carboxylate concentration decreases, indicating 98% starting material consumption. A third electrode is needed for reproducible results [39]. The galvanostatic mode uses a constant current and power supply, making it simpler than potentiostat. It requires only two electrodes and an inexpensive power supply. However, it doesn't control potential, leading to substrates with low redox potential. Continued electrolysis can increase potential, causing over-oxidation. Reporting applied current, electrode dimensions, total charge, or time is crucial for reproducibility [45, 46]. Table 1 shows the various optimizing parameters studied during Kolbe electrolysis.
Electrode selection for efficient Kolbe electrolysis
Catalytic electrodes are gaining attention for their advantages like minimal waste, high atom economy, green reagents, high selectivity, and shorter pathways for multi-step organic reactions. They optimize the electrical double layer, electrode surface properties, and reactant adsorption. A cost-efficient alternative is using affordable materials like nickel (Ni), copper (Cu), cobalt (Co), iron (Fe), palladium (Pd), iridium (Ir), silver (Ag), and gold (Au), stainless steel, sacrificial anodes, boron-doped diamond (BDD), carbon-based electrodes, and conducting carbons [47, 56, 57]. These electrodes produce reactive species by oxidizing or reducing reactants, reducing stable organic complexes, and generating reactive intermediates that further participate in the organic transformation [57]. Recent advancements in Kolbe electrolysis are expected to improve efficiency, selectivity, and industrial applicability [37]. The high performance of Pt makes it a popular choice for anode materials in carboxylic acid conversion [27]. Researchers are seeking innovative electrode materials to increase the surface area, reaction rate, and cost-effectiveness of catalytic electrodes.
Pt-based electrode modifications
Pt and its alloys are superior electrodes due to their high corrosion-resistive electrochemical stability, and excellent HER and oxygen reduction reaction (ORR) catalytic activity, making them a benchmark for various electrochemical energy conversion reactions [56, 85]. Due to these unique features, most studies primarily focus on Pt electrodes, while other self-made electrode materials like thin film Pt, RuO2, IrO2, or BDD have been explored but less commonly used than Pt in large-scale applications [88, 89].
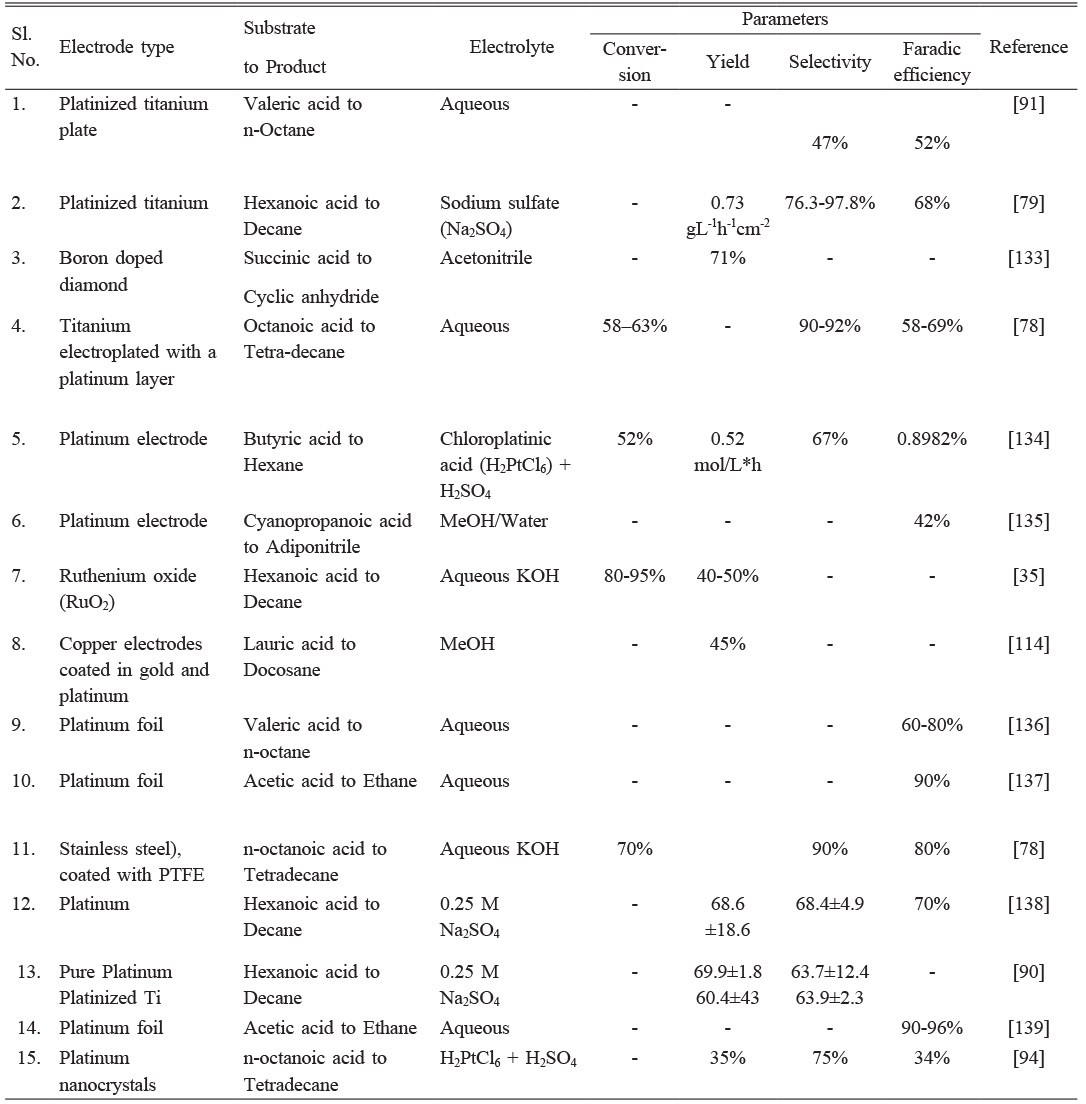
Harnisch proposed platinized titanium (Pt-Ti) electrodes as a cost-effective alternative to platinum bulk electrodes [90, 91], but these anodes are not suitable for alternating current, leading to rapid electrochemical activity loss [14]. Neubert and colleagues have found that Pt-Ti can efficiently convert n-hexanoic acid to n-decane in aqueous solution, achieving a coulombic efficiency of 93.1±6.7%. This results in product selectivity of 66.9 ±0.9%, making Pt-Ti a suitable anode material for Kolbe electrolysis. The degree of Ti surface coverage with Pt was found to be the most important factor, causing a 50% deterioration in coulombic efficiency. Significantly, the process produced 56.7 mL of liquid fuel per mole of n-hexanoic acid, converting to an energy demand of 6.66 kWh and 1.22 € per L, respectively [90]. Recently, a study suggested a self-regulated transition from batch to continuous valeric acid to n-octane using Pt-Ti. The 1 M valeric acid exhibits high selectivity and coulombic efficiency compared to typical batch operations. The study proposes continuous electrosynthesis with product separation, electrolyte recirculation, and online-pH-controlled valeric acid feeding rate, enhancing performance measures at the end of the reaction [91]. Taube et al. study compared the Kolbe electrolysis of myristic acid to produce bio-based hydrophobic paraffin waxes using Pt-Ti as an anode material. Despite no significant performance difference, the low cost of Pt-Ti anodes is advantageous. Further experiments are needed to investigate long-term performance and stability [14]. Yuan et al. developed a method to convert biomass-derived carboxylic acids into fuel-range hydrocarbons using renewable electricity. They used self-supporting core-shell Pt@Ir nanothorns on 3D porous carbon fiber paper anodes, resulting in higher yield and faraday efficiency than commercial Pt/C. The study also found that Pt@Ir nanothorns produced higher tetradecane production, while C7 hydrocarbons (heptane and heptene isomers) were predominant when used as an anode [92].
Nanoparticle-based electrodes coated with Pt significantly improve Kolbe electrolysis performance due to their superior electrochemical properties and catalytic activity. These electrodes facilitate efficient radical formation, improve reaction kinetics and selectivity, and yield desired products. However, the impact of different Pt nanoparticle morphologies on Kolbe electrolysis remains unclear [93, 94]. A low-cost, environmentally friendly method was developed to create self-supporting Pt nanospheres on 3D porous Ti sponge composite electrodes. The Pt-doped Ti sponge (Pt@TS) anode showed enhanced activity and stability for electrocatalytic biofuel production. Both Pt and Pt@TS function as current collectors, but Pt@TS is superior due to its low usage, high surface area, good electrolyte and gas diffusion, and low price compared to pure Pt [93]. Xu and colleagues fabricated Pt nanoparticles (Pt-NPs) on carbon fiber paper to convert n-octanoic acid into n-tetradecane, n-heptane, and n-heptene. The Pt (No.311) plane showed the best selectivity and intrinsic activity for decarboxylation products, especially for Kolbe hydrocarbons. Pt nanothorn (Pt-NT) achieved high selectivity (75%), yield (35%), and faraday efficiency (34%), significantly higher than Pt nanoflowers, Pt nanospheres, and Pt/C [94].
Modified BDD electrodes for enhanced performance
Diamond, a highly abrasive and corrosion-resistant carbon polymorph, has been widely used as an electrode in modern electrochemistry due to its high stability and hydrogen and oxygen overpotentials making it a potential alternative to electrodes with substantial HER, OER, and ORR activity, as it offers the widest stable electrochemical potential window in the aqueous medium [56]. Compton introduced boron-doped diamond i.e. BDD to Kolbe electrolysis as it enhances its electrocatalytic activity and stabilizes organic intermediates [14]. It offers a viable anode material due to its large hydrogen and oxygen overpotentials, excellent chemical stability [14], wider potential windows than Pt [55], and low operational cost making them suitable for harsh electrochemical environments [95-97]. They are emerging next-generation electrode materials for various electrochemistry applications including sensors, organic synthesis, CO2 reduction, ozone water generation, and electrochemiluminescence. Their electrochemical properties are determined by surface termination, surface orientation, and boron doping level [96]. They excel in the complete oxidation and mineralization of persistent organic pollutants, making them versatile for various industrial wastewater treatment applications.
Ashraf and co-investigators found that decarboxylation of acetic acid on BDD electrodes without the OER results in the formation of methanol and methyl acetate. The performance of BDD remains unaffected by current density, concentration, or pH. The selectivity of Pt-modified BDD electrodes to ethane depends on the shape and geometry of Pt particles. Nano-thorn-like Pt particles achieve 40% faradaic efficiency towards ethane, while 3D porous Pt nanoparticles show high selectivity towards the OER. BDD is an ideal substrate for Pt functionalization, offering stability and high-value product formation [98]. A study found that a BDD electrode with current densities of 50 mA/cm2 and 5 mA/cm2 reduced energy usage by 37% for 75% defluorination of perfluorooctanoic acid (PFOA), compared to 50 mA/cm2 alone. Further research on ion-exchange regeneration solutions shows promise for PFOA oxidation [99]. Zeidabadi et al. investigated the use of a BDD anode for remediating common alternatives of PFOA, including perfluorobutanoic acid (PFBA), hexafluoropropylene oxide dimer acid (HFPO-DA, known as GenX and fluorotelomer carboxylic acid (FTCA), in sodium sulfate (Na2SO4) as an electrolyte. Results showed that shorter chains were harder to break down, with PFBA being the most difficult to break down within 120 minutes of electrolysis which follows the order as [PFBA (65.6 ± 5.0%) < GenX (84.9 ± 3.3%) < PFOA (97.9 ± 0.1%) < FTCA (99.4 ± 0.0%)] [100]. Lin and colleagues created nanoporous BDD, a modified BDD electrode with a wide electrochemical window, promising but preventing anodization due to its high stability. High-energy Si (II) ion irradiation forms sp2 defects, allowing BDD anodization and forming nanoporous BDD. Sp2 carbon is pre-formed inside BDD by irradiation. Exposure to electrolytic solution enhances anodic oxidation, potentially causing frontier to intrude into BDD bulk. However, prolonged anodization prevents deeper nanoporous BDD production due to nanopore disappearance, possibly due to electropolishing [101]. Previous research found that BDD electrode surface corrosion occurs in acetic acid solutions, not formic acid solutions, due to methyl radicals formed during Kolbe electrolysis, forming dangling bonds [102]. The study explores Kolbe coupling processes at biphasic Pt and BDD electrodes for electrochemical oxidation of aliphatic carboxylic acids, hexanoic, heptanoic, and lauric acids in the presence of powered ultrasound. The aim was to create an emulsified medium and remove reaction products continuously. The Kolbe dimer product R-R was formed in up to 75% yield with 45% current efficiency for hexanoic acid. The mechanism is explained by a dynamically modified electrode surface, trapping hydrophobic products. Kolbe electrosynthesis is conducted at Pt electrodes and free-standing polycrystalline BDD electrodes to minimize surface erosion [88]. Nonetheless, the question arises as to why boron is doped in diamond and why it cannot act as an electrode alone in organic electrosynthesis. This is because diamond, a metastable carbon allotrope, is not suitable as an electrode material due to its insulator characteristics, such as a bandgap of 5.45 eV at 300 K and high electrical resistivity of 1020 Ω/cm. To reduce ohmic resistance and increase electrical current conductivity, doping with boron creates a semiconductor with a narrow band gap, requiring a large density of boron impurities for sufficient conductivity. Diamond's strong σ-bonds and sp3-hybridized carbon atoms provide outstanding chemical and electrochemical stability. The most striking electrochemical properties of BDD are the large overpotentials in aqueous media for hydrogen and oxygen evolution, allowing the formation of hydroxyl radicals and ozone [103]. To improve electrode quality, input from materials scientists, chemists, and physicists is needed.
RuO2-modified electrodes for improved efficiency
RuO2 electrodes are a promising choice for Kolbe electrolysis and other electrochemical processes due to their high catalytic activity, selectivity towards decarboxylation reaction, unique surface properties, and excellent electrical conductivity [104]. Compared to other metal oxide electrodes, RuO2 shows superior stability under harsh oxidative conditions [105]. They can be optimized for optimal catalytic activity and selectivity, and their performance and durability can be enhanced by doping or alloying with other elements. RuO2 electrodes have been successfully used in organic compound electrochemical synthesis, making them valuable for wastewater treatment and environmental remediation [106-109].
An important work on Kolbe electrolysis investigated the structurally disordered amorphous RuO2 (a-RuO2), which is highly effective in electrocatalytic oxidative decarboxylation of hexanoic acid, producing decane with a yield 5.4 times higher than commercial RuO2. The enhanced product yield is attributed to more efficient carboxylate anions oxidation for alkane dimer formation. This design idea offers a new electrocatalyst candidate for Kolbe electrolysis [110]. On the other hand, the potential of RuO2-coated Ti electrodes has been explored, but the results remain ambiguous due to variations in anode fabrication and reaction conditions [14]. Creusen et al. proposed Ru–Ti-dioxide-coated Ti electrocatalysts as an economical alternative to expensive Pt electrodes for dimerization of ethyl succinate to adipic acid diethyl ester in methanol and aqueous mixtures reactions. Results showed the highest selectivity in methanol (74%). Ti\(RuxTi1–x) O2 electrodes yielded similar current efficiency in methanol up to 75 mol% TiO2 in the coating. Ti\RuO2 anodes also replaced Pt with similar efficiency in aqueous systems. The study confirms an efficient dynamic operation, paving the way for a sustainable chemical industry powered by efficient transient electrocatalytic processes based on windmill energy profiles [111]. The study by Qui et al. presents an electrocatalytic decarboxylation (ECDX) method for converting carboxylic acids into paraffin, olefins, and alcohols using non-Kolbe electrolysis on thin films (TFs). The rate, product selectivity, and current efficiency depend on the electrode. RuO2-TF showed similar activity to Pt foil but lower selectivity to Kolbe products [89]. The research group used RuO2 and Pt nanoparticles to enhance the ECDX of valeric acid into paraffins, olefins, and alcohols. The optimal size for ECDX on RuO2 is around 12 nm, with bulk Pt active for ECDX and Pt NPs only active for oxygen evolution reactions. ECDX current efficiency remains constant on RuO2 NPs. Esterification is preferred at 2.5 V vs. RHE, while Kolbe electrolysis is preferred at 4.5 V vs. RHE [112].
Other electrode systems
As stated, Pt is commonly used as an anode for Kolbe electrolysis due to its high potential stability. Still, other electrode materials like iridium, graphite, carbon, and gold (Au) also catalyze Kolbe radical generation due to their good electrical conductivity and chemical resistivity [32,113-115]. Ahmad et al. found that Pt-coated copper electrodes (Pt-Cu) can efficiently conduct the coupling reaction for the electrochemical oxidation of lauric acid, yielding 45% n-docosane. However, an Au-coated copper electrode (Au-Cu) only yielded 16% n-docosane at a constant current intensity of 85 mA, and no Kolbe product was formed when the current dropped below 85 mA. No further optimization is currently being done, but experimental tests with massive electrodes are ongoing [114]. Interestingly, the Pt-Cu electrodes have a short service life due to surface dissolution effects [114]. This anodic damage may be attributed to the direct dissolution of Pt in the form of Pt ions [116]. Andreev et al. study investigates the conditions for Kolbe cross-electrosynthesis of 10-undecylenic and acetic acids on various catalyst anodes. The main product formed was either hydrocarbon CH3CH2CH(CH3)(CH2)7CH2CH3 (58 wt%), formed by adding three methyl radicals to a vinyl radical, or olefin CH2=CH(CH2)7CH2CH3 (57 wt%), produced by dimerization of vinyl and methyl radicals [113]. In the late 1960s, researchers studied the Kolbe reaction at Au in trifluoroacetic acid solutions. They found similarities between Au and Pt but qualitatively different behavior. Au showed a significant arrest in open-circuit decay, possibly due to adsorbed intermediates. Pt showed the effectiveness of trifluoroacetic anhydride in removing water, with oxide co-adsorption occurring in excess water and oxygen being the main reaction product [32]. The Kolbe electrolysis of n-octanoic acid produced tetradecane in an aqueous alkaline electrolyte. Pt-Ti anodes showed similar results to original Pt-stainless steel electrodes but with better long-term stability and no visible corrosion effects. Insufficiently coated parts showed passivation in Ti, while corrosion and solid iron oxide formation were observed in stainless steel [78].
Commercial titanium dioxide (TiO2) and synthesized TiO2 nanorods decorated by Pt or Au were successfully utilized in the reforming of butyric acid. The study suggests that the coproduction of essential molecules, such as light alkanes and alkenes beyond H2, can be achieved by fine-tuning photocatalyst features [87]. Advanced carbonaceous materials like carbon nanotubes and graphene offer opportunities for electrocatalysis in electro-organic synthesis. Carbon fiber electrodes, with a lower oxidation potential of 1.75 V, significantly mediate C-C coupling and generate radicals and carbocations [117]. Reticulated vitreous carbon (RVC) is a suitable anode for electrochemical C-C activation reactions due to its high surface area and potential zero charge. However, it is unsuitable for an aqueous medium due to its instability in both acidic and basic conditions. This results in many reactions being performed in mild conditions, as organic chemists are not familiar with the stability issues of carbon electrodes [118]. Researchers are exploring methods to enhance carbon electrode stability in an aqueous medium within a mild electrochemical potential window [119], similar to porous bismuth vandate (BiVO4) photoelectrodes, which were performed at <2 V [120].
Ni, a high proton reduction metal, is commonly used as a cathode material in alkaline water electrolyzers as a pristine metal and its compounds [121]. However, due to its high dissolution rate and poor stability, it is not suitable for use in highly acidic environments like sulfuric acid (H2SO4), hydrochloric acid (HCl), and perchloric (HClO4) [122]. A mild organic acid like pivalic acid is an excellent catholyte for Ni electrodes [123] which exhibits significant electrocatalytic activity towards alcohol oxidation in basic solutions, making it suitable for direct methanol fuel cells [124]. Photoelectrochemical alcohol oxidation generates a methoxy radical on ferric oxide (α-Fe2O3), which can be used for organic conversion and the development of new molecules in a sustainable way [125]. Due to the absence of a carboxylate-barrier layer on Au and Ni electrodes, these materials are considered inactive for (non-) Kolbe electrolysis [27]. For anodic substitution reactions including fluorination, methoxylation, acetoxylation, and cyanation of heteroatom compounds containing a sulfur or nitrogen atom, which are fairly similar to Pt, BDD is also shown to be quite successful, followed by glassy carbon electrodes [51].
Active alkaline-earth and d and p-block metals like magnesium, zinc, and aluminum are used as sacrificial electrodes in electro-organic syntheses due to their high oxidation potential. These metals are electrochemically coupled with another material to prevent corrosion, such as stainless steel with magnesium, resulting in a reductive cathode with HER and ORR. This strategy can also be used for electrochemical reduction reactions [56, 126-128]. Adsorption of H+, OH−, and nitrogen atoms in hetero-organic compounds on Pt, Ni, and BDD surfaces is more favorable in terms of energy, enhancing the kinetics of the electro-organic reaction [56, 129, 130]. This anodic oxidation reaction coupled with cathodic reduction reactions like HER and ORR plays a critical role in controlling the rate of various activation reactions [130, 131]. Electro-organic syntheses involve spontaneous reactions of organic intermediates from the counter electrode to form desired products. Hydrogen adsorption occurs via polar hydrogen bonds and aromatic C–H bonds, with hydrogen having a partial positive charge having a higher affinity for electrode surfaces with negative potential. Active intermediates like superoxide and peroxide radicals simplify this process [131]. Table 2 presents an overview of recent synthetic applications of Kolbe electrolysis under various electrode types.
Electrolyte advancements
Importance of green electrolytes
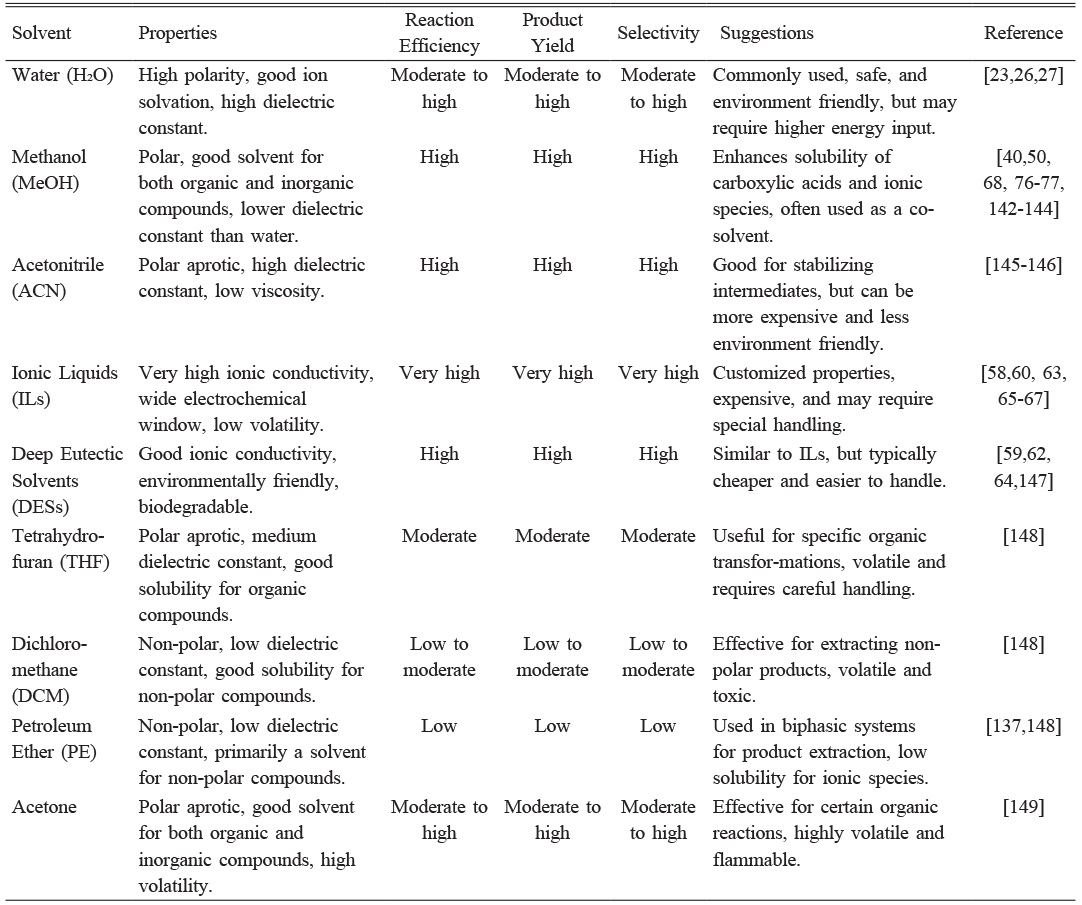
Electrolysis involves subjecting an organic mixture to an electrical current in the presence of solvents, supporting electrolytes, and catalysts. The choice of solvent plays a critical role in the success of the electrolysis and the desired product yield [61, 71]. Kolbe electrolysis commonly uses methanol with a neutral to slightly acidic pH, but nonaqueous solvents like dimethylformamide can be used with gold or platinum electrodes, though they require higher voltage and cooling due to their resistivity [68, 85]. However, aqueous solvents are increasingly favored for their environmental benefits and higher conductivity, which promotes faster reaction rates and supports continuous processes [1, 78]. Aqueous media also facilitate product separation due to the difference in polarity and density between organic products and the solvent, simplifying phase separation and enabling efficient recycling of the aqueous phase [33, 36, 140, 141].
In electro-organic syntheses, supporting electrolytes is essential for reducing cell resistance and ensuring the efficient transport of reagents like reactants and products [61, 71]. These electrolytes are redox stable and provide sufficient electrolytic conductivity (κ), facilitating the passage of electricity throughout the reaction medium. In aqueous solutions, common supporting electrolytes include strong acids, bases, and salts like H2SO4 or potassium hydroxide (KOH), while in organic solvents, they often consist of alkali metals or tetra-alkyl-ammonium salts paired with counter ions such as perchlorates or halides [71]. Organic salts, like pyridinium or quaternary ammonium salts, are typically soluble in organic solvents but can complicate product purification [72, 73]. Some reactants and additives may also serve as ionizable substances, enhancing conductivity while acting as reactants. However, selecting appropriate electrolytes for reactions requiring high electrode potentials presents challenges, and their impact on cost, electrode surfaces, and potential side reactions must be carefully considered [61]. Table 3 highlights various solvent systems used in Kolbe electrolysis.
Next-generation electrolytes
Ionic liquids (ILs)
Ionic liquids (ILs) are highly attractive for high-voltage reactions, such as Kolbe electrolysis, due to their low volatility, high thermal and chemical stability, and excellent ionic conductivity [107, 150]. With a wide electrochemical window and negligible vapor pressure, ILs are ideal for sustained electrochemical operations in both laboratory and industrial settings, offering enhanced safety [150].
Applications in cosmetics and pharmaceuticals, such as Dierker's fatty acid dimerization and Bradin's conversion of sugars and triglycerides into fuels, underscore the importance of electrolyte choice. Bradin et al. recommended ILs to improve efficiency in Kolbe electrolysis, particularly for acid transformations where water sensitivity is an issue. Alkane products derived from triglycerides could further serve as fuels or base stock oils [108]. Moreover, ILs have shown success in decarboxylation processes, such as the synthesis of hydroxyapatite, decarboxylation of itaconic acid, and the catalytic conversion of cyclic carbonates to epoxides, showcasing their versatility beyond traditional Kolbe electrolysis [109-111].
Deep eutectic solvents (DES)
Deep eutectic solvents (DESs) are a sustainable and eco-friendly alternative to traditional solvents in chemical processes. Their low volatility, tunable solubility, and ease of preparation make them ideal for green chemistry applications, particularly in decarboxylation of fatty acids, crucial for hydrocarbon production and biofuels [147, 151]. DES is a type of IL, composed of a mixture of two or more components, forming a eutectic mixture with a lower melting point than individual components. Common components include quaternary ammonium salts like choline chloride and hydrogen bond donors like urea, glycerol, or organic acids [59]. Exploring DES for its environment-friendly properties and ability to dissolve a wide range of substrates is highly recommended. The limited solubility of phenolic acid decarboxylase enzyme in industrial processes has been overcome by DES i.e. Choline chloride (ChCl)-based eutectic solvents with 0-50% water content. The choice of solvent also influences substrate acceptance, as DES strongly favors the conversion of caffeic acid, which is only poorly converted in aqueous media [64].
Electrolysis cell configurations in Kolbe reaction
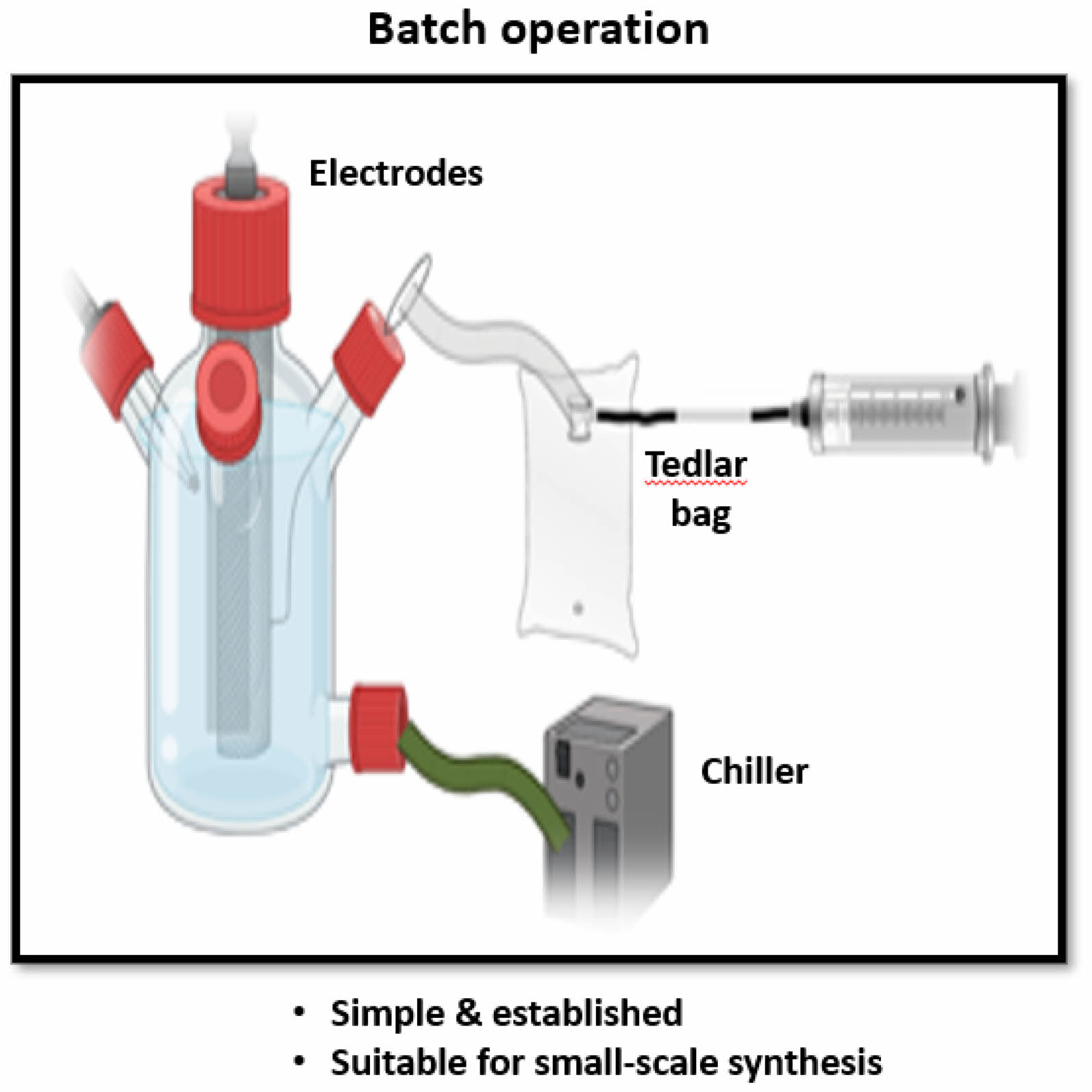
Advances in cell design and configuration are crucial for enhancing the performance of electrochemical synthesis [52]. The Kolbe reaction is often performed in simple setups, such as an undivided cell, beaker, or tube, with two platinum electrodes and a power supply with high anode potential and current density [10, 153]. Cooling equipment, such as ice baths or chillers, is used for temperature control [154]. modern electro-organic synthesis, undivided cells are common and straightforward, involving two parallel plates immersed in an electrolytic solution. For higher productivity, multiple parallel plates can be used. Although undivided cells are simple, they risk reverse reactions at the counter electrode, leading to non-productive processes. This issue can be mitigated by using a sacrificial reaction, such as hydrogen gas evolution at the cathode [39]. The more complex H-cell, or divided cell, prevents undesired reactions at the counter electrode by separating the anode and cathode compartments with a porous material or membrane. This design is particularly useful in paired electrolysis and simplifies product isolation [39]. The quasi-divided cell design, which combines the advantages of divided and undivided cells, uses a large working electrode with low current density and a small counter electrode with high current density, preventing the electrolysis of starting materials at the counter electrode [39, 46,74].
Modern cell design: Continuous flow cells and Microreactors
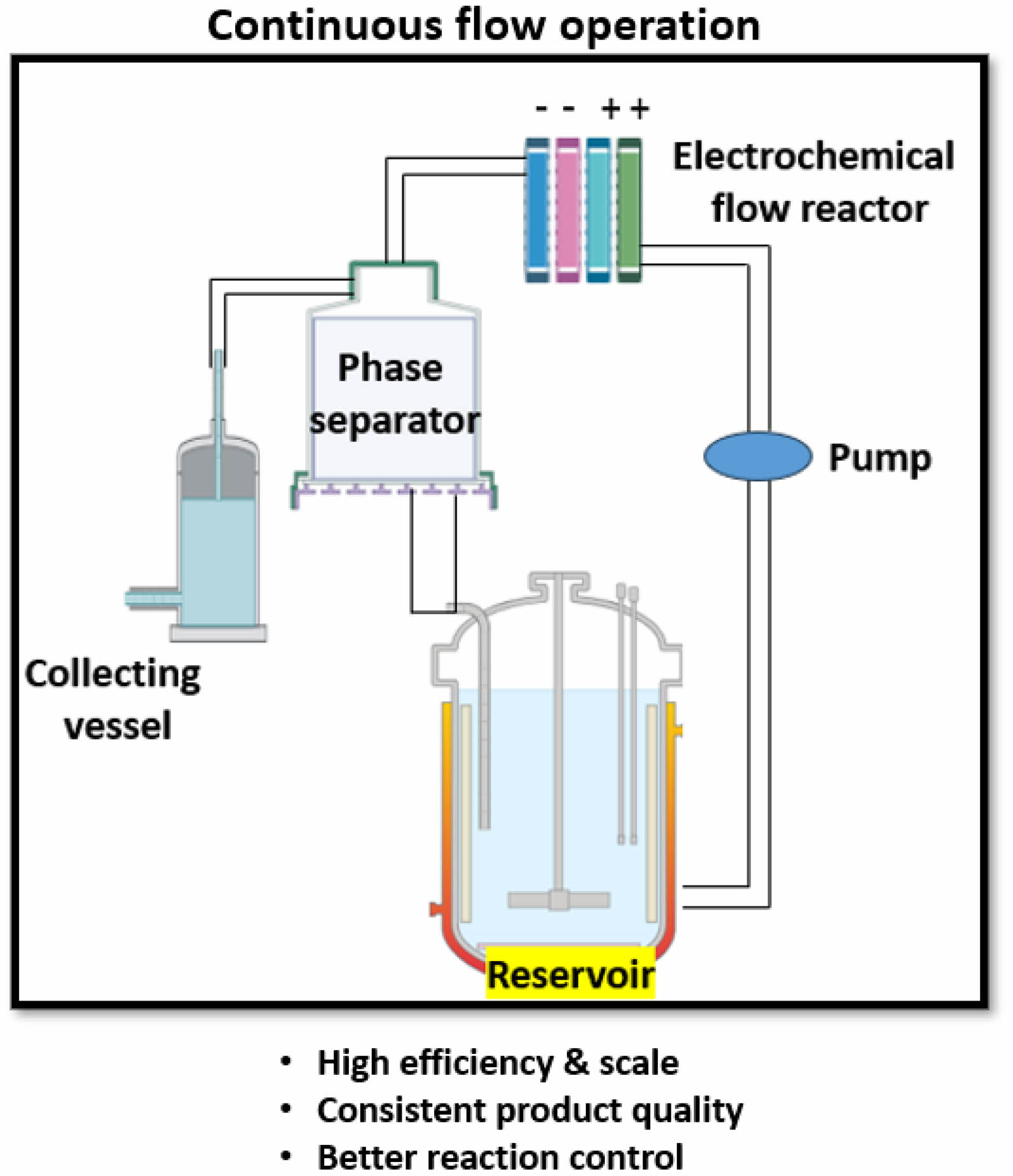
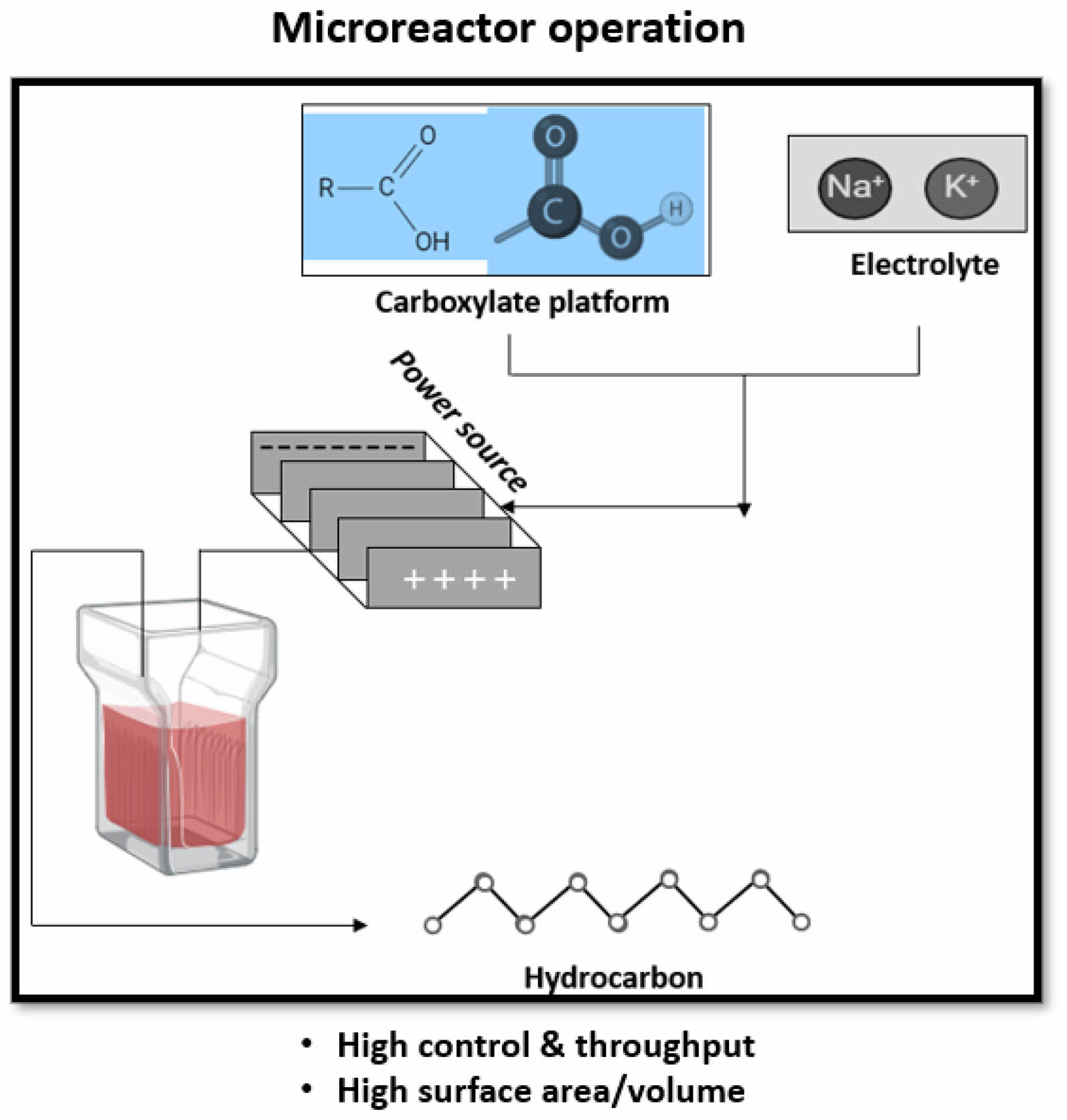
Over the past two decades, flow technology has significantly impacted the manufacturing of chemical entities, particularly pharmaceuticals, blurring the lines between chemistry and chemical engineering. This trend suggests that flow chemistry and related technologies will play a crucial role in modern chemical manufacturing, laying the foundation for the 4th industrial revolution [83]. Continuous flow technologies improve efficiency, product quality, reduce reaction times, and allow consistent reaction conditions (Fig. 3) [83]. On the other hand, microreactors provide precise control over reaction parameters, enhance safety, and enable high-throughput screening of reaction conditions, (Fig. 4) [84].
Electrolysis under flow conditions, where a reactant solution is pumped between electrodes at short distances, reduces cell resistance and allows for lower concentrations of added electrolytes compared to batch electrolysis. This method eliminates the risk of overoxidation, a major drawback of batch reactor setup (Fig. 5). Various research groups have developed electrochemical flow cells for diverse reactions, and some designs have been commercialized, making flow electrolysis an attractive alternative in organic synthesis [80-82, 155]. Continuous flow reactors, operating under steady-state conditions, provide improved control over reaction parameters, enhancing reaction efficiency and selectivity. They also facilitate safer handling of reactive chemicals by processing smaller volumes, making them suitable for industrial-scale production. In Kolbe electrolysis, continuous flow reactors maintain optimal reaction conditions and ensure consistent product quality, critical for industrial applications [78, 91, 155]. For example, continuous production of ethane from acetic acid using Pt or BDD electrodes yields high selectivity under controlled conditions [98]. Dos Santos et al. demonstrated that using a flow reactor with reduced residence time increased the selectivity of Kolbe products, such as n-octane, from 51% to 81% [140].
Microreactors are particularly advantageous for Kolbe electrolysis, enabling the rapid screening of reaction conditions and facilitating high throughput, which leads to high yields of higher alkanes from fatty acids under controlled conditions. These systems are modular, safe, and easily scalable for both research and production. Microreactors are particularly advantageous for Kolbe electrolysis, enabling rapid screening of reaction conditions and high throughput, leading to high yields of higher alkanes from fatty acids under controlled conditions [78, 79]. A pilot plant for continuous Kolbe electrolysis of fatty acids was constructed using a modular electrochemical microreactor from Fraunhofer-Institut für Mikrotechnik und Mikrosysteme (Fraunhofer IMM). This system integrates continuous electrosynthesis, separation of the organic phase from the aqueous electrolyte, electrolyte recycling, and automated product reconcentration. The electrodes, featuring integrated heat exchanger channels, were fabricated through selective laser melting and electroplated with Pt. The microreactor design includes a plane electrode arranged between two microstructured electrodes, housed in a reactor volume of 0.64 mL per electrochemical cell, with Polytetrafluoroethylene (PTFE) sealings to prevent leakage [78, 141]. Therefore, addition of aqueous KOH is essential for stabilizing the electrolyte's pH and reducing potassium bicarbonate (KHCO3) concentration, achieving stable conversion and Kolbe selectivity within 75 minutes. To optimize electrolysis times, KHCO3 removal before reconcentration was suggested. Significant studies on Kolbe flow-cell electrolysis have been conducted by the Schröder and Wirth groups, who successfully suppressed the undesired Hofer-Moest product 1-butanol during the Kolbe dimerization of valeric acid to n-octane. The Wirth group also demonstrated a custom-designed microreactor for the dimerization of aromatic carboxylic acids, while Brown and Pletcher explored the dimerization of biogenic monomethyl adipate [86,140,156-158]. Additionally, Kurig et al. focused on implementing Kolbe chemistry in flow electrochemistry, achieving a 75% optical density in the semi-batch dimerization of levulinic acid, which surpassed batch results. They successfully converted the biobased substrate 3-hydroxy decanoic acid (3-HDA) in a single-pass setup, achieving 98% selectivity and a five-fold increase in hourly production [159].
A study has successfully transitioned valeric acid to n-octane from a batch to a continuous reaction using MicroFlowCell (ELECTROCELL, Denmark). The process, which uses PTFE flow frames and Pt-Ti plate electrodes, addresses key green chemistry rules, improving waste prevention, and energy efficiency, and allowing for easy control through online monitoring. The design also enhances selectivity, coulombic efficiency, passive product separation, and electrolyte reconditioning, ensuring a sustainable and efficient process [91]. A two-chamber electrochemical flow cell was used for scaling and process engineering of Kolbe electrolysis, specifically for converting n-hexanoic acid. The cell had a chamber volume of 100 mL and electrodes equipped with a turbulence promoter mesh. Both electrodes were Pt-Ti with a liquid contact surface area of 100 cm2. The electrolysis performance was exceptional, with a maximum n-decane production rate of 0.73 g L−1h−1 cm−1, selectivity between 76.3% and 97.8%, and coulombic efficiencies up to 68% [79]. A study presents a cascading continuous stirred tank reactor (CSTR) with individual cell potential control, demonstrating a balance between high selectivity and throughput for electrochemistry in electrochemical synthesis. The IKA ElectraSyn 2.0 was used for small-scale reactions, offering greater modularity and access to commercially available electrode materials. Future efforts will focus on improving mass transport through impeller geometry optimization and characterization to increase throughput, reducing the barrier to implementing scale-up electrochemical reactions [160]. Finally, automation and smart reactors are transforming the industry by integrating sensors and automated control systems in continuous flow and microreactors, ensuring real-time monitoring and parameter adjustment, and using artificial intelligence (AI) and machine learning (ML) to predict optimal conditions, and dynamically adjust parameters, and diagnose issues [161-163]. In conclusion, continuous flow and microreactor technologies are poised to revolutionize Kolbe electrolysis for hydrocarbon synthesis. They offer enhanced efficiency, scalability, and control while integrating advanced materials, automation, and sustainable practices for a greener future.
Future directions: Electrifying chemical synthesis with Kolbe electrolysis
The chemical industry, which produces essential products like polymers, fuels, fertilizers, and pharmaceuticals, is a cornerstone of modern civilization [164]. However, the sector's heavy reliance on fossil fuels for both energy and raw materials has historically contributed to significant greenhouse gas (GHG) emissions [165]. The ongoing shift towards electrification presents a transformative opportunity to realign the chemical industry with sustainable development goals [166]. Electrification involves replacing fossil fuel-based processes with renewable electricity, driving chemical reactions through electrosynthesis and electrochemical processes, and integrating renewable energy sources such as wind, solar, and hydropower to reduce thermal energy dependence [167, 168]. The integration of renewable electricity into chemical production processes is crucial for eliminating the industry's carbon footprint, thereby contributing to global climate goals. Recent technological advancements and cost reductions in renewable energy have accelerated the decarbonization of the power sector [169], but pathways to decarbonize the industrial sector responsible for 24% of global GHG emissions in 2019 remain more complex. Scalable and sustainable technologies are needed to decouple economic growth from rising emissions [170]. Among these technologies, Kolbe electrolysis offers significant potential for the electrification of chemical processes. This electrochemical process has been used for various industrial applications, including the commercial production of sebacic acid, bio-lubricants, and even biodiesel from biomass [171, 172]. What makes Kolbe electrolysis particularly relevant in the context of electrification is its ability to operate under mild conditions, reduce reliance on hydrogen, and integrate seamlessly with renewable energy sources, thereby enhancing its environmental benefits (Fig. 6).
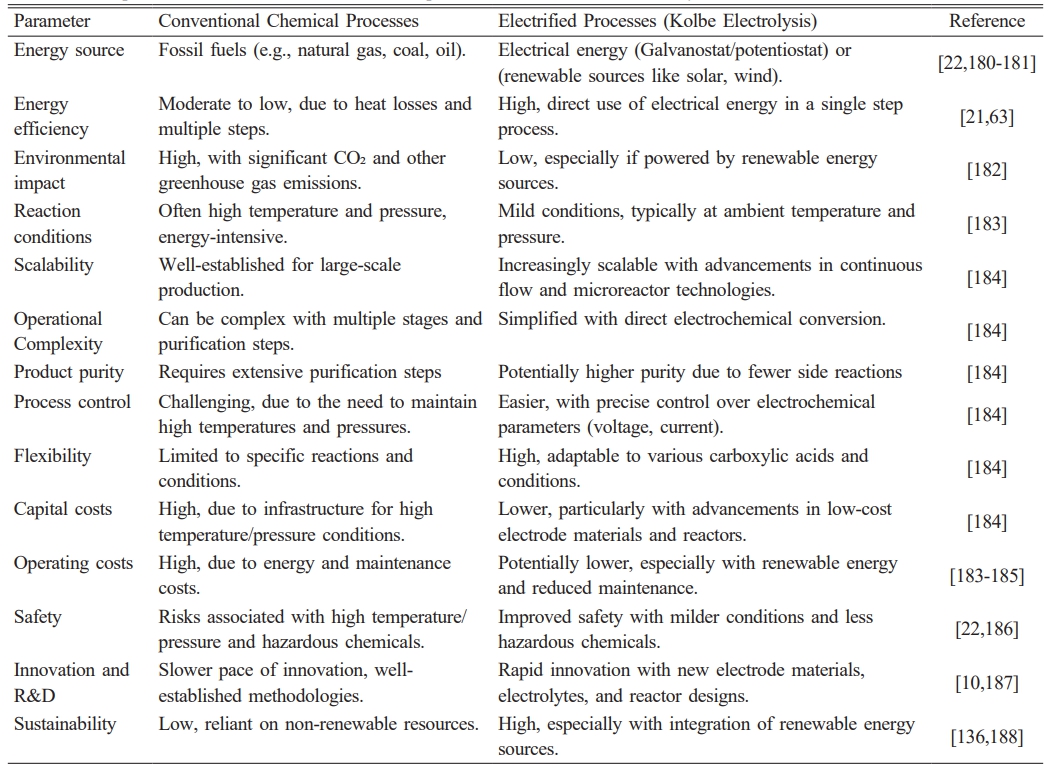
Kolbe electrolysis also plays a role in decarbonizing the chemical industry by reducing the need for carbon-intensive feedstocks. For example, it enables the commercial production of sebacic acid from adipic acid, through the decarboxylative dimerization of carboxylic acids [173], conversion of biomass into biodiesel [171, 172], and carbon electrode grafting [174, 175]. This makes it a promising technology to produce bio-lubricants and other high-value chemicals while minimizing environmental impacts. The process is efficient in producing high-quality hydrocarbons and can be adapted for use with renewable energy, further lowering its carbon footprint [171]. However, despite its advantages, several challenges remain. The scale-up of Kolbe electrolysis is one of the most pressing issues, as is the recovery of dissolved electrode materials and the separation of electrolytes. Addressing these challenges will be critical to making the process economically viable on an industrial scale. Additionally, the market price of bio-lubricant products may influence the overall economic feasibility of this technology [171, 172, 174, 176]. Table 4 presents a comparative analysis of various parameters between conventional and electrified processes in the chemical industry.
Electro catalytic processes are used to drive biological or biochemical transformations, offering a sustainable approach to energy production and chemical synthesis [177-179]. Kolbe electrolysis is a promising reaction for the transformation of biologically derived chemicals to value added products. Intensive research and development will be essential to overcome the obstacles and unlocking the full potential of Kolbe electrolysis. Innovations in reactor design, the use of ionic liquids as electrolytes, and the development of more efficient electrocatalysts could significantly enhance the process's performance and make it highly competitive in the production of sustainable chemicals. The ongoing collaboration between leading chemical companies and research institutions highlights the importance of this technology in driving the future of industrial chemistry toward greener and more eco-friendly solutions. Kolbe electrolysis, when integrated with renewable energy and aligned with the goals of electrification, represents a promising avenue for the sustainable transformation of the chemical/biochemical industry. Table 5 provides a comprehensive list of various research organizations and institutes involved in electrochemical processes, potentially including Kolbe electrolysis.
|
Fig. 1 Schematic overview of Kolbe electrolysis setup showing the production of hydrocarbons using carboxylic acid as substrate under mild conditions. |
|
Fig. 2 Schematic representation of the mechanism of non-Kolbe electrolysis. |
|
Fig. 3 Schematic representation of continuous flow operation for Kolbe electrolysis. |
|
Fig. 4 Schematic representation of microreactor operation for Kolbe electrolysis. |
|
Fig. 5 Schematic representation of batch operation for Kolbe electrolysis. |
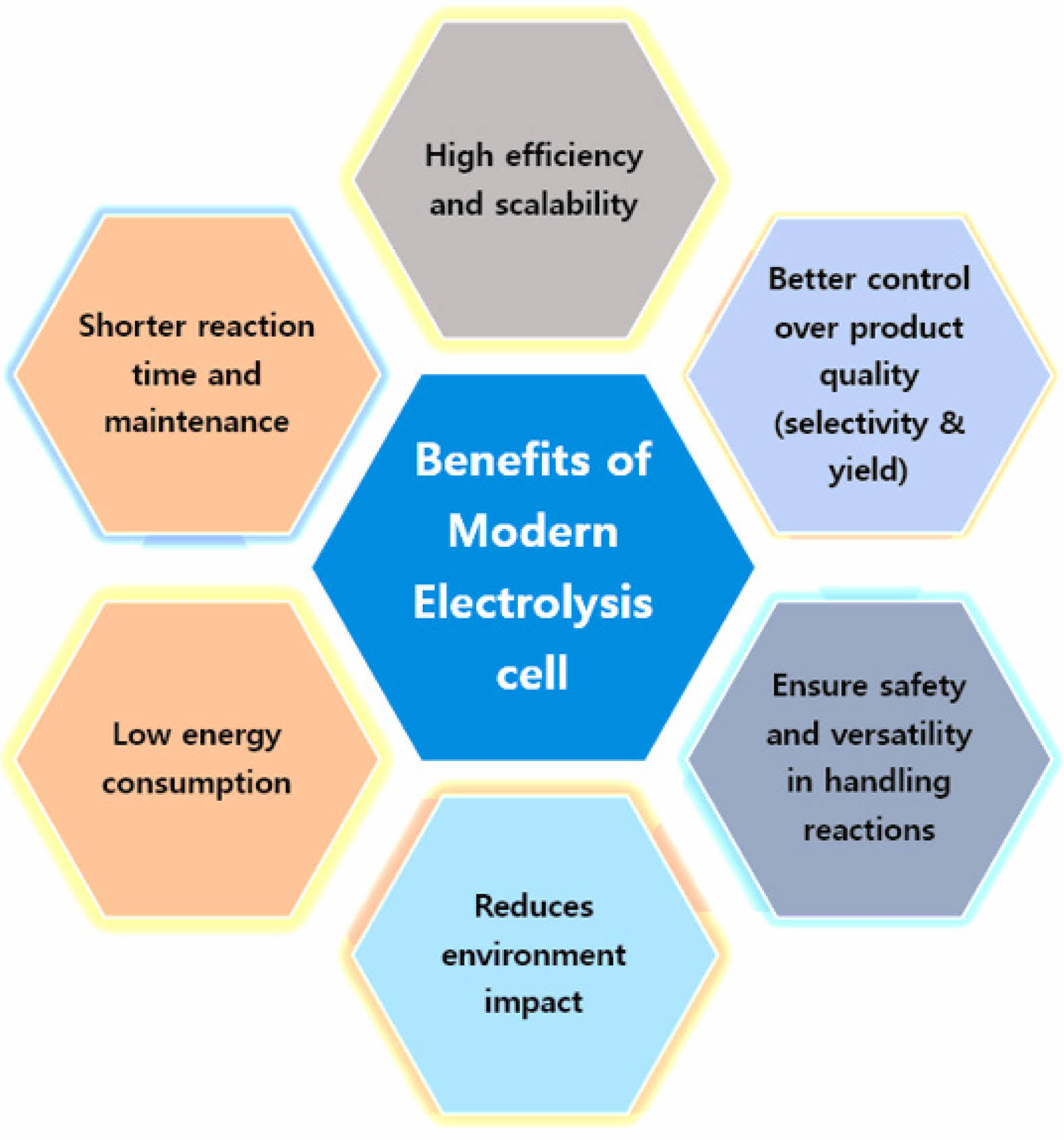
|
Fig. 6 An outlines showing the benefits of modern electrolysis cells. |
|
Table 1 Optimization parameters and their effects on product yield and selectivity. |
|
Table 2 Overview of recent studies on Kolbe electrolysis using various electrode types. |
|
Table 4 Comparison of conventional vs. electrified processes in the chemical industry. |
|
Table 5 List chemical industries, academic institutes, and startups working in the field of electrochemistry. |

Kolbe electrolysis, a traditional method for decarboxylating carboxylic acids to produce hydrocarbons, has seen significant advancements and is now a crucial technique in modern chemical synthesis. The process involves optimizing reaction conditions like temperature, pressure, and cell design, as well as choosing the right electrolytes and solvents. The mode of operation also significantly impacts the efficiency and selectivity of electrochemical reactions. Recent applications of Kolbe electrolysis demonstrate its versatility and potential for synthesizing a wide range of hydrocarbons. This process aligns with green chemistry goals, especially when integrated with renewable energy sources, supporting the electrification of the chemical industry. The transition to continuous flow and microreactor technologies enhances the control, scalability, and safety of electrochemical processes. The future of Kolbe electrolysis is promising, driven by innovations in electrode materials, the introduction of novel electrolytes, and advanced reactor designs. As the chemical industry evolves towards more sustainable practices, Kolbe electrolysis remains a powerful and adaptable tool, contributing significantly to the future of sustainable chemical production.
This work was supported by Korea Environment Industry & Technology Institute (KEITI) through Development of Demonstration Technology for energy conversion using waste resources Program, funded by Korea Ministry of Environment (MOE) (2022003480001).
The authors announce no conflict of interest.
- 1. F. Harnisch and U. Schröder, Chem. Electro. Chem. 6[16] (2019) 4126-4133.
-

- 2. Y.H. Budnikova, E.L. Dolengovski, M.V. Tarasov, and T.V. Gryaznova, J. Solid State Electrochem. 28[3] (2024) 659-676.
-
- 3. G. Hilt, Curr. Opin. Electrochem. (2024) 101425.
-
- 4. S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe, and S.R. Waldvogel, Angew. Chem. Int. Ed. 57[21] (2018) 6018-6041.
-
- 5. S. Imeni, A. Makarem, and R. Javahershenas, Asian J. Org. Chem. 12[8] (2023) e202300303.
-
- 6. S. Garg, H.S. Sohal, D.S. Malhi, M. Kaur, K. Singh, A. Sharma, V. Mutreja, D. Thakur, and L. Kaur, Curr. Org. Chem. 26[10] (2022) 899-919.
-
- 7. F.C. Walsh, C.P. de León, C. Kerr, S. Court, and B.D. Barker, Surf. Coat. Technol. 202[21] (2008) 5092-5102.
-
- 8. P. Ragupathy, S.D. Bhat, and N. Kalaiselvi, WIREs Energy Environ. 12[2] (2023) e464.
-
- 9. L. Mais, N. Melis, A. Vacca, and M. Mascia, Environ. Sci. Water Res. Technol. 10[2] (2024) 399-407.
-
- 10. N. Sbei, S. Aslam, and N. Ahmed, React. Chem. Eng. 6[8] (2021) 1342-1366.
-
- 11. M. Sugiya and H. Nohira, Bull. Chem. Soc. Jpn. 73[3] (2000) 705-712.
-
- 12. H.J. Schäfer, Eur. J. Lipid Sci. Technol. 114[1] (2012) 2-9.
-
- 13. S. Lateef, S.R. Mohan, and S.R. Reddy, Tetrahedron Lett. 48[1] (2007) 77-80.
-
- 14. C. Taube, A. Fischer, and M. Beyer, Chem. Cat. Chem. (2024) e202400628.
-
- 15. A.V. Shtelman and J.Y. Becker, J. Org. Chem. 76[11] (2011) 4710-4714.
-
- 16. L. Catacuzzeno, A. Michelucci, and F. Franciolini, Biomolecules 14[6] (2024) 684.
-
- 17. I.D. Ayodeji, S.A. Ayodeji, B.J. Abiodun, and A. Odun, in “Innovative developments of materials for economic diversification” (NIMACON, 2020) p.18.
- 18. O. Smutok and E. Katz, J. Solid State Electrochem. 28[3] (2024) 683-710.
-
- 19. M. Faraday, Ann. Phys. 109[31‐34] (1834) 481-520.
-
- 20. H. Kolbe, Justus Liebigs Ann. Chem. 69[3] (1849) 257-294.
-
- 21. D. Klüh, W. Waldmüller, and M. Gaderer, Clean Technol. 3[1] (2021) 1-8.
-
- 22. A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes, and S.R. Waldvogel, Angew. Chem. Int. Ed. 57[20] (2018) 5594-5619.
-
- 23. J.A. Stapley and J.N. BeMiller, Carbohydr. Res. 342[3-4] (2007) 610-613.
-
- 24. M. Galicia, M.A. González-Fuentes, D.P. Valencia, and F.J. González, J. Electroanal. Chem. 672 (2012) 28-33.
-
- 25. S. Payamifar, L. Behrouzi, and A.P. Marjani, Arabian J. Chem. (2024) 105822.
-
- 26. M. Li and X. Cheng, Isr. J. Chem. 64[1-2] (2024) e202300067.
-
- 27. M.O. Nordkamp, T. Ashraf, M. Altomare, A.C. Borca, P. Ghigna, T. Priamushko, S. Cherevko, V.A. Saveleva, C. Atzori, A. Minguzzi, and X. He, Surf. Interfaces 44 (2024) 103684.
-
- 28. B. Xu, D. Li, Q. Zhao, S. Feng, X. Peng, and P.K. Chu, Coord. Chem. Rev. 502 (2024) 215609.
-
- 29. E. Santillan-Jimenez and M. Crocker, J. Chem. Technol. Biotechnol. 87[8] (2012) 1041-1050.
-
- 30. M.C. Leech and K. Lam, Acc. Chem. Res. 53 (2020) 121-134.
-
- 31. F.J. Holzhauser, J.B. Mensah, and R. Palkovits, Green Chem. 22 (2020) 286-301.
-
- 32. A.K. Vijh and B.E. Conway, Chem. Rev. 67 (1967) 623-664.
-
- 33. P. Nilges, T.R. dos Santos, F. Harnisch, and U. Schröder, Energy Environ. Sci. 5 (2012) 5231-5235.
-
- 34. S. Liu, N. Govindarajan, H. Prats, and K. Chan, Chem. Catal. 2[5] (2022) 1100-1113.
-
- 35. S. Wang, D. Ren, Y. Du, M. Zhang, N. Zhang, Y. Sun, and Z. Huo, Carbon Resour. Convers. 6[4] (2023) 287-297.
-
- 36. C. Urban, J. Xu, H. Sträuber, T.R. dos Santos Dantas, J. Mühlenberg, C. Härtig, L.T. Angenent, and F. Harnisch, Energy Environ. Sci. 10[10] (2017) 2231-2244.
-
- 37. K. Hiromori, Y. Konno, K. Katagami, A. Takahashi, and N. Shibasaki-Kitakawa, J. Chem. Eng. Jpn. 57[1] (2024) 2332621.
-
- 38. J.E. Sanderson, P.F. Levy, L.K. Cheng, and G.W. Barnard, J. Electrochem. Soc. 130[9] (1983) 1844.
-
- 39. N. Amri, Electroorg. Synth. Automated Flow Platform (Doctoral Dissertation, Cardiff University, 2021).
- 40. S.D. Ross, M. Finkelstein, and E.J. Rudd, in “Anodic Oxidation: Organic Chemistry: A Series of Monographs” (Elsevier Press, 2013) Chapter 6.
- 41. P. Hapiot and C. Lagrost, Chem. Rev. 108[7] (2008) 2238-2264.
-
- 42. K. Aasberg-Petersen, E. Stenby, and A. Fredenslund, Ind. Eng. Chem. Res. 30[9] (1991) 2180-2185.
-
- 43. A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino, and D.F. Rivas, Joule 4[3] (2020) 555-579.
-
- 44. M.N. Salehmin, T. Husaini, J. Goh, and A.B. Sulong, Energy Convers. Manag. 268 (2022) 115985.
-
- 45. O. Hammerich and B. Speiser, in “Organic Electrochemistry” (CRC Press, 2016).
- 46. G. Hilt, Chem. Electro. Chem. 7[2] (2020) 395-405.
-
- 47. K.I. Ataka, T. Yotsuyanagi, and M. Osawa, J. Phys. Chem. 100[25] (1996) 10664-10672.
-
- 48. K. Pecková, J. Musilová, and J. Barek, Crit. Rev. Anal. Chem. 39[3] (2009) 148-172.
-
- 49. A. Li, W. Duan, J. Liu, K. Zhuo, Y. Chen, and J. Wang, Sci. Rep. 8[1] (2018) 13141.
-
- 50. G.H. de Kruijff, and S.R. Waldvogel, Chem. Electro. Chem. 6[16] (2019) 4180-4183.
-
- 51. H. Nojima, T. Shimoda, S. Inagi, and T. Fuchigami, Isr. J. Chem. 64[1-2] (2024) e202300025.
-
- 52. L. Cong, Y. Wu, N. Lin, X. Li, F. Liu, F. Han, J. Yang, C. Wang, and H. Lin, Chem. Eng. J. 480 (2024) 148331.
-
- 53. X.L. Sun, C.X. Xia, Y. Ren, Y.J. Li, Z.Q. Cao, and L.G. Meng, Org. Chem. Front. 11[8] (2024) 2189-2194.
-
- 54. S. Abdullaev, D. Singh, M.N. Al-Delfi, A. Kumar, Q.H. Aziz, A. Elawady, M.A. Al-Anber, A.H. Al-Rubaye, A. Ali, and N. Ahmad, Appl. Organomet. Chem. 38[5] (2024) e7425.
-
- 55. H. He, Z. Lin, W. Guo, H. Zhang, H. Li, and W. Huang, Sep. Purif. Technol. 212 (2019) 802-821.
-
- 56. P.E. Karthik, I. Alessandri, and A. Sengeni, J. Electrochem. Soc. 167 (2020) 125503.
-
- 57. A. Iqbal, J. Zai, and Y. Zhang, in “Nanomaterials in Electro-organic Synthesis” (RSC Press, 2022) p.1-25.
-
- 58. J.S. Choi, F.S. Simanjuntak, J.Y. Oh, K. Im Lee, S.D. Lee, M. Cheong, H.S. Kim, and H. Lee, J. Catal. 297 (2013) 248-255.
-
- 59. E.L. Smith, A.P. Abbott, and K.S. Ryder, Chem. Rev. 114[21] (2014) 11060-11082.
-
- 60. M. Kathiresan and D. Velayutham, Chem. Commun. 51[99] (2015) 17499-17516.
-
- 61. C. Stang and F. Harnisch, Chem. Sus. Chem. 9[1] (2016) 50-60.
-
- 62. P. Makoś, A. Przyjazny, and G. Boczkaj, J. Chromatogr. A 1570 (2018) 28-37.
-
- 63. H. Qi, Y. Ren, S. Guo, Y. Wang, S. Li, Y. Hu, and F. Yan, ACS Appl. Mater. Interfaces 12[1] (2019) 591-600.
-
- 64. A.K. Schweiger, N. Ríos-Lombardía, C.K. Winkler, S. Schmidt, F. Morís, W. Kroutil, J. González-Sabín, and R. Kourist, ACS Sustain. Chem. Eng. 7[19] (2019) 16364-16370.
-
- 65. F.J. Holzhäuser, J.B. Mensah, and R. Palkovits, Green Chem. 22[2] (2020) 286-301.
-
- 66. S. Pang, H. An, X. Zhao, and Y. Wang, Chin. J. Chem. Eng. 67 (2024) 9-15.
-
- 67. Y. Li, L. Wang, Y. Cao, S. Xu, P. He, H. Li, and H. Liu, RSC Adv. 11[23] (2021) 14193-14202.
-
- 68. R.G. Woolford, Can. J. Chem. 40[9] (1962) 1846-1850.
-
- 69. C. Reufer, T. Lehmann, C. Weckbecker, inventors; Degussa GmbH, assignee, U.S. Patent App. US 10/546,135 (2006) 20 July.
- 70. A. Fischer, H. Pütter, inventors; BASF SE, assignee, U.S. Patent US 7,192,512 (2007) 20 March.
- 71. A.J. Bard, G. Inzelt, and F. Scholz, in “Electrochemical Dictionary” (Springer, 2012) p.1.
-
- 72. T. Fuchigami, M. Atobe, and S. Inagi, in “Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices” (John Wiley & Sons, 2014).
-
- 73. E.J. Horn, B.R. Rosen, and P.S. Baran, ACS central Sci. 2[5] (2016) 302-308.
-
- 74. B.K. Malviya, E.C. Hansen, C.J. Kong, J. Imbrogno, J. Verghese, S.M. Guinness, C.A. Salazar, J.N. Desrosiers, C.O. Kappe, and D. Cantillo, Org. Process Res. Dev. 28[3] (2024) 790-797.
-
- 75. E. Santillan‐Jimenez and M. Crocker, J. Chem. Technol. Biotechnol. 87[8] (2012) 1041-1050.
-
- 76. L. Brakha and J.Y. Becker, Electrochim. Acta 77 (2012a) 143-149.
-
- 77. L. Brakha and J.Y. Becker, Electrochim. Acta 59 (2012b) 135-139.
-
- 78. N. Baumgarten, B.J. Etzold, J. Magomajew, and A. Ziogas, Chem. Open 11[10] (2022) e202200171.
-
- 79. L.F. Rosa, K. Röhring, and F. Harnisch, Fuel 356 (2024) 129590.
-
- 80. D. Pletcher, R.A. Green, and R.C. Brown, Chem. Rev. 118[9] (2017) 4573-91.
-
- 81. M. Elsherbini and T. Wirth, Acc. Chem. Res. 52[12] (2019) 3287-96.
-
- 82. T. Noël, Y. Cao, and G. Laudadio, Acc. Chem. Res. 52[10] (2019) 2858-69.
-
- 83. N.C. Neyt and D.L. Riley, React. Chem. Eng. 6[8] (2021) 1295-326.
-
- 84. D.K. Nguyen, F. Cameli, P. Dimitrakellis, and D.G. Vlachos, Ind. Eng. Chem. Res. 63[20] (2024) 9008-9017.
-
- 85. V.R. Stamenkovic, B.S. Mun, M. Arenz, K.J. Mayrhofer, C.A. Lucas, G. Wang, P.N. Ross, and N.M. Markovic, Nature Mater. 6[3] (2007) 241-247.
-
- 86. G. Creusen, F.J. Holzhäuser, J. Artz, S. Palkovits, and R. Palkovits, ACS Sustainable Chem. Eng. 6[12] (2018) 17108-17113.
-
- 87. G. Scandura, M. Sajjad, N. Singh, G. Palmisano, and J. Rodríguez, Appl. Catal. A Gen. 624 (2021) 118321.
-
- 88. J.D. Wadhawan, F.J. Del Campo, R.G. Compton, J.S. Foord, F. Marken, S.D. Bull, S.G. Davies, D.J. Walton, and S. Ryley, J. Electroanal. Chem. 507[1-2] (2001) 135-143.
-
- 89. Y. Qiu, J.A. Lopez-Ruiz, U. Sanyal, E. Andrews, O.Y. Gutiérrez, and J.D. Holladay, Appl. Catal. B: Environ. 277 (2020) 119277.
-
- 90. K. Neubert, M. Schmidt, and F. Harnisch, Chem. Sus. Chem. 14[15] (2021) 3097-3109.
-
- 91. P. Drögemüller, T. Stobbe and U. Schröder, Chem. Sus. Chem. 17[2] (2024) e202300973.
-
- 92. G. Yuan, L. Wang, X. Zhang, R. Luque, and Q. Wang, ACS Sustain. Chem. Eng. 7[21] (2019) 18061-18066.
-
- 93. G. Yuan, L. Wang, X. Zhang, R. Luque, and Q. Wang, Green Chem. 22[2] (2020) 525-531.
-
- 94. S. Xu, X. Niu, G. Yuan, Z. Wang, S. Zhu, X. Li, Y. Han, R. Zhao, and Q. Wang, ACS Sustain. Chem. Eng. 9[15] (2021) 5288-5297.
-
- 95. J.V. Macpherson, Phys. Chem. Chem. Phys. 17[5] (2015) 2935-2949.
-
- 96. Y. Einaga, Acc. Chem. Res. 55[24] (2022) 3605-3615.
-
- 97. S. Palkovits and R. Palkovits, Chem. Ing. Tech. 91[6] (2019) 699-706.
-
- 98. T. Ashraf, A.P. Rodriguez, B.T. Mei, and G. Mul, Faraday Discuss. 247 (2023) 254-269.
-
- 99. M. Ensch, C.A. Rusinek, M.F. Becker, and T. Schuelke, Water Environ. J. 35[1] (2021) 158-165.
-
- 100. F.A. Zeidabadi, E.B. Esfahani, R. Moreira, S.T. McBeath, J. Foster, and M. Mohseni, Environ. Res. 246 (2024) 118103.
-
- 101. C. Lin, Y. Maeda, K. Murase, and K. Fukami, Electrochem. Commun. 149 (2023) 107473.
-
- 102. T. Kashiwada, T. Watanabe, Y. Ootani, Y. Tateyama, and Y. Einaga, ACS Appl. Mater. Interfaces 8[42] (2016) 28299-28305.
-
- 103. S. Lips and S.R. Waldvogel, Chem. Electro. Chem. 6[6] (2019) 1649-1660.
-
- 104. Y. Zhang, Z. Zhang, Z. Yu, A. Addad, Q. Wang, P. Roussel, S. Szunerits, and R. Boukherroub, ACS Appl. Mater. Interfaces 15[50] (2023) 58345-58355.
-
- 105. A. Goryachev, M.E. Pascuzzi, F. Carla, T. Weber, H. Over, E.J. Hensen, and J.P. Hofmann, Electrochim. Acta 336 (2020) 135713.
-
- 106. L.Å. Näslund, Á.S. Ingason, S. Holmin, and J. Rosen, J. Phys. Chem. C 118[28] (2014) 15315-15323.
-
- 107. N. Ullah, I. Ali, M. Jansen, and S. Omanovic, Can. J. Chem. Eng. 93[1] (2015) 55-62.
-
- 108. C.P. Rhodes, J.F. Godinez Salomon, L.A. Albiter, K.O. Bailey, Z.G. Naymik, F.A. Ospina Acevedo, and P.B. Balbuena, ECS Meet. Abstr. 242 (2022) 1649.
-
- 109. F.A. Ospina Acevedo, P.B. Balbuena, J.F. Godinez Salomon, L.A. Albiter, K.O. Bailey, Z.G. Naymik, and C.P. Rhodes, ECS Meet. Abstr. 242 (2022) 1670.
-
- 110. C. Wang, K. Liu, Y. Jin, S. Huang, and J. Chun‐Ho Lam, Chem. Sus. Chem. 16[16] (2023) e202300222.
-
- 111. G. Creusen, F.J. Holzhäuser, J. Artz, S. Palkovits, and R. Palkovits, ACS Sustain. Chem. Eng. 6[12] (2018) 17108-17113.
-
- 112. Y. Qiu, J.A. Lopez-Ruiz, G. Zhu, M.H. Engelhard, O.Y. Gutiérrez, and J.D. Holladay, Appl. Catal. B Environ. 305 (2022) 121060.
-
- 113. V.N. Andreev, V.I. Bykov, V.A. Grinberg, A.G. Dedov, A.S. Loktev, N.A. Mayorova, I.I. Moiseev, and A.A. Stepanov, Russ. J. Electrochem. 49 (2013) 216-220.
-
- 114. H. Ahmad, R. Pelosato, I.N. Sora, and F. Fontana, Chem. Eng. Trans. 98 (2023) 105-110.
- 115. S. Abdullaev, D. Singh, M.N. Al‐Delfi, A. Kumar, Q.H. Aziz, A. Elawady, M.A. Al‐Anber, A.H. Al‐Rubaye, A. Ali, and N. Ahmad, Appl. Organomet. Chem. 38[5] (2024) e7425.
-
- 116. J. Ranninger, P. Nikolaienko, K.J.J. Mayrhofer, and B.B. Berkes, Chem. Sus. Chem. 15 (2022) e202102228.
-
- 117. C.L. Arnold, K.M. Beggs, D.J. Eyckens, F. Stojcevski, L. Servinis, and L.C. Henderson, Compos. Sci. Technol. 159 (2018) 135-141.
-
- 118. A. Ambrosi and M. Pumera, Chem. Eur. J. 22[1] (2016) 153-159.
-
- 119. H. Tomiyasu, H. Shikata, K. Takao, N. Asanuma, S. Taruta, and Y.Y. Park, Sci. Rep. 7 (2017) 45048.
-
- 120. W.A. Swansborough-Aston, A. Soltan, B. Coulson, A. Pratt, V. Chechik, and R.E. Douthwaite, Green Chem. 25[3] (2023) 1067-1077.
-
- 121. M. Chhetri, S. Sultan, and C.N. Rao, Proc. Natl. Acad. Sci. 114[34] (2017) 8986-8990.
-
- 122. M. Wickramasinghe and I.Z. Kiss, J. Electrochem. Soc. 163[14] (2016) H1171.
-
- 123. N. Sauermann, T.H. Meyer, C. Tian, and L. Ackermann, J. Am. Chem. Soc. 139[51] (2017) 18452-18455.
-
- 124. M.A. Rahim, R.A. Hameed, and M.W. Khalil, J. Power Sources 134[2] (2004) 160-169.
-
- 125. C.A. Mesa, A. Kafizas, L. Francàs, S.R. Pendlebury, E. Pastor, Y. Ma, F. Le Formal, M.T. Mayer, M. Grätzel, and J.R. Durrant, J. Am. Chem. Soc. 139[33] (2017) 11537-11543.
-
- 126. J. Chaussard, J.C. Folest, J.Y. Nedelec, J. Perichon, S. Sibille, and M. Troupel, Synthesis 1990[05] (1990) 369-381.
-
- 127. C. Rousseau, F. Baraud, L. Leleyter, and O. Gil, J. Hazard. Mater. 167[1-3] (2009) 953-958.
-
- 128. W. Xu, J. Song, J. Sun, Y. Lu, and Z. Yu, ACS Appl. Mater. Interfaces 3[11] (2011) 4404-4414.
-
- 129. K. Pecková, J. Musilová, and J. Barek, Crit. Rev. Anal. Chem. 39[3] (2009) 148-172.
-
- 130. A. Li, W. Duan, J. Liu, K. Zhuo, Y. Chen, and J. Wang, Sci. Rep. 8[1] (2018) 13141.
-
- 131. C. Amatore and A.R. Brown, J. Am. Chem. Soc. 118[6] (1996) 1482-1486.
-
- 132. A.J. Bard, G. Inzelt, and F. Scholz, in “Electrochemical Dictionary” (Springer, 2012).
-
- 133. J. Schneider, A. P. Häring, and S. R. Waldvogel, Chem. Eur. J. 30[30] (2024) e202400403.
-
- 134. H. Lee, A Study of Kolbe Electrolysis Using High-Energy Facet Pt Electrode, Doctoral Dissertation (Hanyang University, 2024).
- 135. R. Mathison, E. Rani, M.K. Patel, A.L. Cerrato, C.K. Bloomquist, and M.A. Modestino, Chem. Catal. 4[5] (2024) 100998.
-
- 136. N. Teetz, D. Holtmann, F. Harnisch, and M. Stöckl, Angew. Chem. 134 (2022) e202210596.
-
- 137. M.O. Nordkamp, B. Mei, R. Venderbosch, and G. Mul, Chem. Cat. Chem. 14[16] (2022) e202200438.
-
- 138. K. Neubert, M. Hell, M. Chávez Morejón, and F. Harnisch, Chem. Sus. Chem. 15[21] (2022) e202201426.
-
- 139. H. Soucie, M.O. Nordkamp, E. Faegh, M. Elam, G. Mul, and W. Mustain, ECS Meet. Abstr. 240 (2021) 75.
-
- 140. T.R. dos Santos, P. Nilges, W. Sauter, F. Harnisch, and U. Schröder, RSC Adv. 5[34] (2015) 26634-26643.
-
- 141. A. Ziogas, H. Pennemann, and G. Kolb, Electrocatalysis 11 (2020) 432-442.
-
- 142. E. Steckhan, in “Electrochemical synthesis: bond formation at anode and cathode” (Springer,1990).
-
- 143. E. Klocke, A. Matzeit, M. Gockeln, and H. J. Schäfer, Chem. Ber. 126[7] (1993) 1623-1630.
-
- 144. C. Capello, U. Fischer, and K. Hungerbühler, Green Chem. 9[9] (2007) 927-934.
-
- 145. J.F. Wilshire, Aust. J. Chem. 16[3] (1963) 432-439.
-
- 146. M.M. Baizer and J.H. Stocker, J. Electrochem. Soc. 121[3] (1974) 134C.
-
- 147. S. Joarder, D. Bansal, H. Meena, N. Kaushik, J. Tomar, K. Kumari, I. Bahadur, E.H. Choi, N.K. Kaushik, and P. Singh, J. Mol. Liq. 376 (2023) 121355.
-
- 148. M. Quertenmont, I. Goodall, K. Lam, I. Markó, and O. Riant, Org. Lett. 22[5] (2020) 1771-1775.
-
- 149. L. Eberson, J. Org. Chem. 27[7] (1962) 2329-2331.
-
- 150. M. Kathiresan and D. Velayutham, Chem. Commun. 51[99] (2015) 17499-17516.
-
- 151. P. Makoś, A. Przyjazny, and G. Boczkaj, J. Chromatogr. A 1570 (2018) 28-37.
-
- 152. K. Scott, Dev. Chem. Eng. Miner. Process. 1[2-3] (1993) 71-117.
-
- 153. A.K. Yadav, A. Jain, and R.A. Misra, Electrochim. Acta 27[4] (1982) 535-540.
-
- 154. N. Ahad and A. de Klerk, Fuel 211 (2018) 415-419.
-
- 155. D. Cantillo, Curr. Opin. Electrochem. 44 (2024) 101459.
-
- 156. K. Arai, K. Watts, and T. Wirth, Chem. Open 3 (2014) 23-28.
-
- 157. K. Watts, W. Gattrell, and T. Wirth, Beilstein J. Org. Chem. 7 (2011) 1108-1114.
-
- 158. R. A. Green, R. C. D. Brown, and D. Pletcher, J. Flow Chem. 6 (2016) 191-197.
-
- 159. N. Kurig, J. Meyers, F.J. Holzhauser, S. Palkovits, and R. Palkovits, ACS Sustain. Chem. Eng. 9 (2020) 1229-1234.
-
- 160. J.D. Griffin, K.C. Harper, S. Velasquez Morales, W.H. Morrill, W. I. Thornton, D. Sutherland, and B.A. Greiner, Org. Process Res. Dev. 28 (2024) 1877-1885.
-
- 161. C.P. Breen, A.M. Nambiar, T.F. Jamison, and K.F. Jensen, Trends Chem. 3 (2021) 373-386.
-
- 162. P. Nikolaienko and K.J. Mayrhofer, Curr. Opin. Electrochem. 35 (2022) 101103.
-
- 163. D.Z. Lin, G. Fang, and K. Liao, in “Machine Learning in Molecular Sciences” (Springer, 2023) p. 227-275.
-
- 164. A. Jess and P. Wasserscheid, in “Chemical Technology: From Principles to Products” (John Wiley & Sons, 2020).
- 165. W. R. Moomaw, Energy Policy 24 (1996) 951-968.
-
- 166. Z.J. Schiffer and K. Manthiram, Joule 1 (2017) 10-14.
-
- 167. P. De Luna, C. Hahn, D. Higgins, S.A. Jaffer, T.F. Jaramillo, and E. H. Sargent, Science 364 (2019) eaav3506.
-
- 168. I. Ganesh, Renew. Sustain. Energy Rev. 59 (2016) 1269-1297.
-
- 169. D.S. Mallapragada, Y. Dvorkin, M.A. Modestino, D.V. Esposito, W.A. Smith, B.M. Hodge, M.P. Harold, V.M. Donnelly, A. Nuz, C. Bloomquist, and K. Baker, Joule 7 (2023) 23-41.
-
- 170. P.R. Shukla, J. Skea, R. Slade, A. Al Khourdajie, R. Van Diemen, D. McCollum, M. Pathak, S. Some, P. Vyas, R. Fradera and M. Belkacemi, in Climate Change 2022: Mitigation of Climate Change, Contribution of Working Group III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change (2022) 9781009157926.
-
- 171. C.K. Ho, K.B. McAuley, and B.A. Peppley, Renew. Sustain. Energy Rev. 113 (2019) 109261.
-
- 172. G. Yuan, C. Wu, G. Zeng, X. Niu, G. Shen, L. Wang, X. Zhang, R. Luque, and Q. Wang, Chem. Cat. Chem 12 (2020) 642-648.
-
- 173. M. Seko, Y. Yomiyama, and T. Isoya, Chem. Econ. Eng. Rev. 11 (1979) 48-50.
-
- 174. C. P. Andrieux, F. Gonzalez, and J. M. Savéant, J. Am. Chem. Soc. 119 (1997) 4292-4300.
-
- 175. D. Belanger and J. Pinson, Chem. Soc. Rev. 40 (2011) 3995-4048.
-
- 176. H. Wendt, H. Vogt, G. Kreysa, M. Kolb, G.E. Engelmann, J.C. Ziegler, H. Goldacker, K. Juttner, U. Galla, H. Schmieder, and E. Steckhan, in “Ullmann's Encyclopedia of Industrial Chemistry” (Wiley, 2003) p.1.
-
- 177. J. Lee, J. Chun, O. Choi, and B.-I. Sang, J. Ceram. Process. Res. 21, [5] (2020) 602-608.
-
- 178. S. Kang, A. K. Mathew, A. Abraham, O. Choi, and B.-I. Sang, J. Ceram. Process. Res. 23, [6] (2022) 853-861.
-
- 179. A. Udayan, S. Kang, and B.-I Sang, J. Ceram. Process. Res. 24[1] (2023) 29-39.
-
- 180. J. Egerer, N. Farhang-Damghani, V. Grimm, and P. Runge, Appl. Energy 358 (2024) 122485.
-
- 181. L. T. Angenent, I. Casini, U. Schröder, F. Harnisch, and B. Molitor, Energy Environ. Sci. 17 (2024) 3682-3699.
-
- 182. A. Hassan, S. Z. Ilyas, A. Jalil, and Z. Ullah, Environ. Sci. Pollut. Res. 28 (2021) 21204-21211.
-
- 183. P. J. Megia, A. J. Vizcaíno, J. A. Calles, and A. Carrero, Energy Fuels 35 (2021) 16403-16415.
-
- 184. P. G. Levi and J. M. Cullen, Environ. Sci. Technol. 52 (2018) 1725-1734.
-
- 185. M. Jiang, Y. Cao, C. Liu, D. Chen, W. Zhou, Q. Wen, H. Yu, J. Jiang, Y. Ren, S. Hu, and E. Hertwich, Nat. Commun. 15 (2024) 3854.
-
- 186. L. R. López, P. Dessì, A. Cabrera-Codony, L. Rocha-Melogno, B. Kraakman, V. Naddeo, M. D. Balaguer, and S. Puig, Sci. Total Environ. 856 (2023) 159088.
-
- 187. G. Centi and S. Perathoner, Catal. Today 387 (2022) 216–223.
-
- 188. L. Puigjaner, M. Pérez-Fortes, A. Somoza-Tornos, and A. Espuña, Front. Energy Res. 9 (2021) 780533.
-
 This Article
This Article
-
2024; 25(6): 1087-1104
Published on Dec 31, 2024
- 10.36410/jcpr.2024.25.6.1087
- Received on Nov 1, 2024
- Revised on Nov 30, 2024
- Accepted on Dec 5, 2024
Services
Shared
Correspondence to
- Byoung-In Sang
-
Department of Chemical Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 04763, Republic of Korea
Tel : +82-2-2220-2328 - E-mail: biosang@hanyang.ac.kr
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr