





- Influence of sulfuric acid doping on sintering behaviors of nano-Yttria powder and production of transparent ceramics
Xin Zhang, Jiao He*, Jingbao Lian, Xue Zhang and Mingxia Lei
School of Mechanical Engineering, Liaoning Petrochemical University, Fushun, 113001, P.R. China
This article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
The present study investigated the effects of sulfuric acid doping on the structure, morphology, and sintering behavior of nano Y2O3 powders. The sintering kinetics of the resultant compacts during the initial sintering stage were assessed, using constant rates of heating (CRH). Compared to the undoped Y2O3 powder, the powder doped with sulfuric acid exhibited improved nano-scale uniformity in particle size and enhanced homogeneity in particle dispersion. The introduction of sulfuric acid as a dopant significantly boosted the densification rate of Y2O3 at temperatures exceeding ~1350 °C. This addition decreased the activation energy required for sintering and transformed the diffusion mechanism, shifting it from grain boundary (GB) diffusion to volume diffusion (VD) during the initial sintering stage. Microstructural and optical characterization of as-fabricated ceramics were employed to further evaluate the sintering behavior of powders. When being sintered at 1500 ℃ in vacuum, a homogeneous microstructure was observed in the ceramic derived from the doped Y2O3 powder, whereas large amounts of pores were presented in the undoped sample. At a sintering temperature of 1700 ℃ for 5 h, the undoped ceramic appeared opaque, while the doped sample exhibited remarkable transparency, achieving a transmittance of approximately 74% at a wavelength of 2000 nm.
Keywords: Sintering behaviour, Transparent ceramics, Y2O3 nano-powders, Sulfuric acid.
Yttria (Y2O3) transparent ceramics possess various outstanding physical, mechanical, and chemical characteristics, such as high-temperature stability, robust corrosion resistance, and excellent optical transparency. These exceptional qualities of Y2O3 transparent ceramics have led to their extensive development and employment in various optical applications, including missile infrared domes, semiconductor components, host materials for scintillators, as well as solid lasers, etc. [1-4].
In order to achieve superior transparency in sintered materials, internal porosity must be eliminated entirely in the final sintering stage [5, 6]. More recent works have been focused on the utilization of nanopowders with high sintering activity [2], as well as various sintering techniques such as hot pressing (HP) [7], hot isostatic pressing (HIP) [8], spark plasma sintering (SPS) [9, 10], or their combinations [11]. Furthermore, doping with sintering additives containing HfO2 [12], La2O3 [13], ThO2 [14], LiF [15], and ZrO2 [16] has been investigated as a potential method to enhance the transparency of ceramics. Fu et al. [17] obtained transparent Y2O3 by a combination of air presintering at 1500 ℃ and HIP sintering at 1550 ℃ under 200 MPa pressure, using ZrO2 as sintering additives. ZrO2 suppressed grain boundary mobility while promoted pore pinning, causing highly dense ceramics. The in-line transmittance at 1100 nm (1.0 mm thick) wavelength reached 81.4%. Although pressure-assisted sintering methods are effective for producing highly transparent ceramics, these methods have substantial limitations, particularly when producing components with large sizes and complex geometrical shapes. Additionally, the high-pressure operating environment also poses some risks and high costs. Recently, Park et al. [13] fabricated transparent Y2O3 ceramics via SPS method, adding 3 mol% La2O3. The sintered sample showed excellent transmittance of 79.44% at a wavelength of 600 nm. However, SPS technology has the drawback of high equipment costs. Unfortunately, the use of a significant amount of sintering additives can alter the stoichiometry of the material, leading to the formation of a secondary phase that may separate from the boundary, ultimately resulting in a negative impact on transparency of ceramics [18].
Hence, to produce high-quality Y2O3 transparent ceramics by pressureless sintering, preparing nano-powder endowed with well sinterability is widely accepted as a critical process [19]. Among numerous preparation methods, the precipitation synthesis method is preferred for preparing ceramic powders due to its high yield and minimal equipment requirements [20]. Many studies on precipitation techniques for producing sinterable Y2O3 powders often utilize additives, including SO42−-ions, as dopants to achieve dispersion and regulate the shape of the resulting Y2O3 powders [21-29]. Zhang et al. synthesized nano-Y2O3 powders with weak agglomeration by calcining hydroxyl-carbonate precursors obtained from a spray co-precipitation technique. The mean particle size of the powders calcined at 1250 ℃ was approximately 140 nm. (NH4)2SO4 was used as a regulator during the process. The Y2O3 ceramic, subjected to vacuum sintering at 1800 ℃ for 8 h, demonstrated an in-line transmittance of 78.7% at a wavelength of 1064 nm [29]. Our earlier study [30] examined the impact of introducing doped SO42--ions to a commercially available Y2O3 powder. This process involved treating the starting powders with sulfuric acid. Loosely agglomerated and sinterable powder upon calcination was obtained from the powders treated with 7 mol% sulfuric acid and calcined at 1100 ℃ for 4 h. This was due to the presence of a single layer coverage of SO42−-ions on the surfaces of the particles, which inhibited the formation of hard agglomerates by facilitating the movement of particles during the calcination process, leading to particle coarsening.
Moreover, it is noted that the exploration of the sintering process kinetics of ceramic powder, especially at the initial sintering stage, holds significant importance for improving sintering technology [31]. Numerous researchers have previously examined the initial sintering of various ceramic powders, and various sintering-rate equations utilized for kinetic analysis during the initial sintering stage have been reported [32-37]. By utilizing sintering models, it is feasible to determine the mechanism, the activation energy of the process, and various other physical constants of the system. Among them, the assessment of quantitative densification during the initial sintering stage, executed under constant rate of heating (CHR), is widely acknowledged. Koji Matsui et al. studied the kinetic behavior of the initial sintering stage in Y-TZP powders doped and undoped Al2O3 [38]. The results showed that a minor quantity of Al2O3 increased the densification rate at the initial sintering stage, owing to a decrease in the activation energy of volume diffusions (VD) and a change of diffusion mechanism from grain-boundary diffusion mechanism (GBD) to (VD). Subsequently, he reported that the elevated Y2O3 concentration in fine zirconia powder led to a increase in the activation energy of GBD [39]. Prior endeavors have mostly concentrated on investigating the impact of synthetic conditions on the properties of Y2O3 powders. Nevertheless, the sintering kinetics of Y2O3 ceramic nano-powders, vital for producing densely packed ceramics, have yet to receive sufficient attention.
The present study focused on obtaining Y2O3 nano-powders with varying morphologies and particle size distribution (PSD) through calcination, both with and without the addition of sulfuric acid. This study employed constant rates of heating (CRH) to examine the influence of sulfuric acid doping on the sintering kinetics of nano-Y2O3 powders during the initial sintering stage. Additionally, the microstructure and optical properties of as-prepared ceramics that were vacuum-sintered were thoroughly analyzed to detect the sintering behaviors of the powders.
Preparation process
The starting material for this research was commercially available Y2O3 powder, a 99.99% purity level, acquired from Huizhou Ruier Rare Chemical Hi-Tech Co., Ltd., Huizhou, China. This powder exhibited a Brunauer-Emmett-Teller (BET) surface area of 31.5 m2/g. Two distinct batches of Y2O3 powder were prepared: one undoped and the other doped with 7 mol% sulfuric acid. The sulfuric acid concentration was calculated based on the molar percentage relative to the Y2O3 powder. Both powders groups underwent ball-milling, drying, and sieving through a 200-mesh nylon sieve. For comprehensive details, refer to our prior publication [30]. In atmospheric conditions, the so-obtained powders were calcined at 1100 ℃ for 4 h. Subsequently, they were uniaxially pressed into green compacts at a pressure of 100 MPa and then cold isostatically pressed at 200 MPa. The green compacts finally underwent sintering at 1500 ℃ and 1700 ℃ for 5 h in a vacuum furnace equipped with a molybdenum heating element (VSF-7, Shenyang, China).
Characterization
High-resolution transmission electron microscopy (TEM), employing a JEM-2100F microscope from JEOL, Tokyo, Japan, was utilized to examine the morphologies of the calcined Y2O3 powders.
Phase identification of calcined Y2O3 powders was carried out through X-ray diffractometry, employing a Philips Model PW3040/60 instrument based in Eindhoven, The Netherlands. Operating conditions for the XRD included a current of 40 mA, alongside the use of graphite monochromatic CuKα radiation (λ=0.15406 nm). Particle size distribution was analyzed utilizing LA-920 particle size analyzer (HORIBA, Japan), with distilled water serving as the dispersing medium.
The densification of powder compacts was evaluated in an oxygen environment (flow rate: 20 mL/min) via dilatometry using a thermal mechanical analyzer (SETSYS Evolution 2400, Setaram, Caluire, France). The analyzer maintained a constant heating rate from 2.5 ℃/min to 20 ℃/min, reaching a maximum temperature of 1600 ℃.
The green density of the green compacts was calculated using the geometric measurement technique. The density of the sintered bodies was determined through the Archimedes method, employing deionized water as the immersion medium at ambient temperature. Subsequently, the relative density was calculated by comparing the actual density of the sintered bodies with the theoretical density of Y2O3.
The in-line transmittance of the polished ceramic specimens, which were 1.5 mm thick, was evaluated utilizing a UV/Visible/NIR spectrophotometer (Model Lambda 750S, Perkin Elmer, CT, US).
A scanning electron microscope was employed to analyze the fracture surfaces of vacuum-sintered specimens. In order to prevent surface charge effects, especially at high magnification and voltage, prior to observation, the samples were uniformly coated with a gold layer.
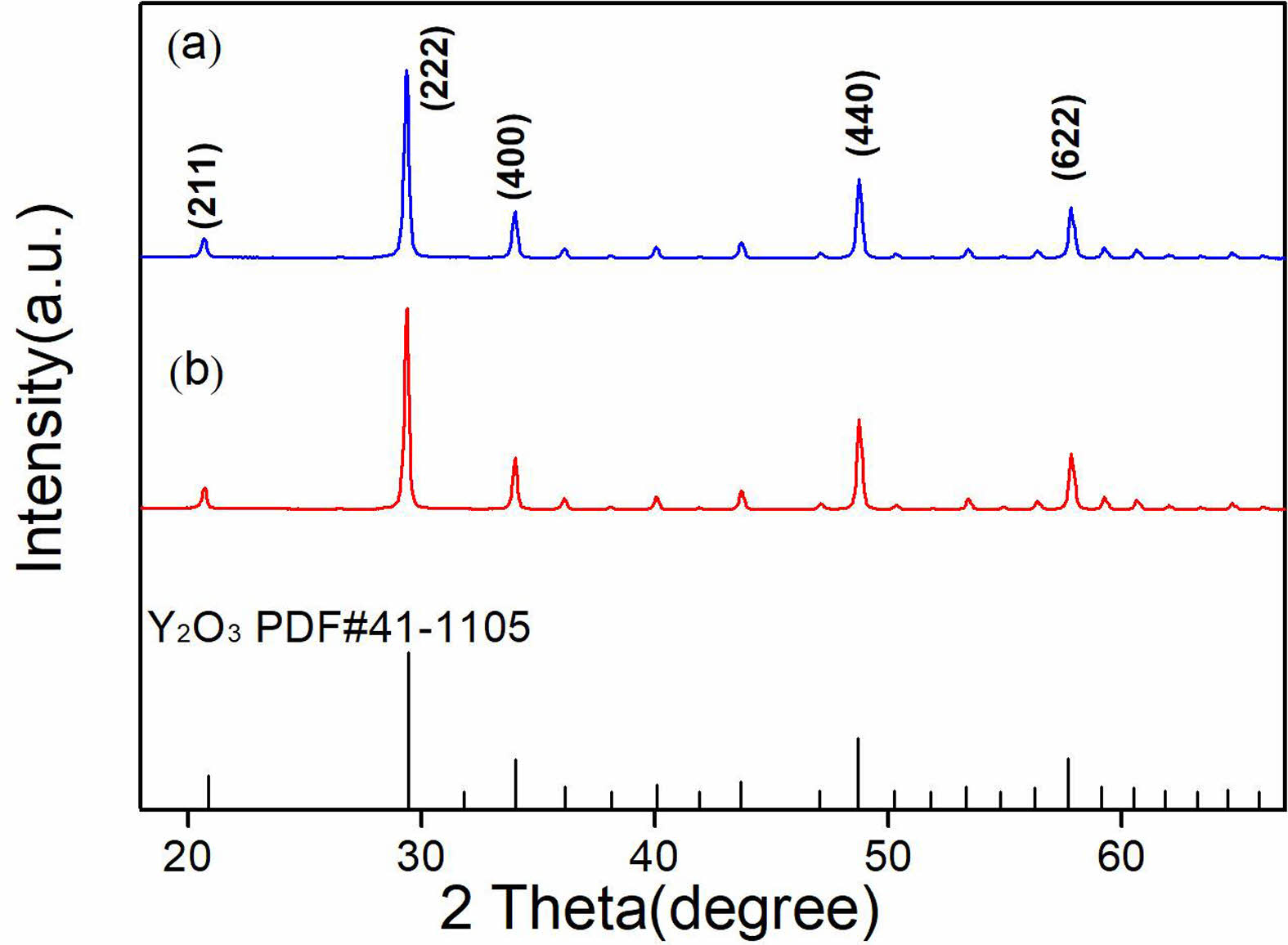
Figure 1 presents the XRD patterns of calcined Y2O3 powders, undoped (a) and doped (b) with sulfuric acid. The XRD patterns exhibited a similarity, corresponding to the cubic yttria phase (JCPDS#41-1105). Both characteristic diffraction peaks were sharp, indicating a high degree of crystallinity. Additionally, the results suggest that including sulfuric acid did not affect the formation of the Y2O3 phase.
Figure 2 illustrates the morphologies of Y2O3 powders that were calcined at 1100 ℃ for 4 h. A comparison is made between the undoped powder (Fig. 2(a)) and the powder doped with sulfuric acid (Fig. 2(b)). Figure 2(a) shows that the powders mainly consisted of severely agglomerated and irregular particles, with sharp edges and flat surfaces. The average particle size (dTEM) was approximately 107 nm. However, when sulfuric acid was used, the resulting powders were less-aggregated and more evenly dispersed. As illustrated in Fig. 2(b), the powders exhibited an average particle size (dTEM) of approximately 90 nm. Additionally, most particles exhibited round surfaces, indicating a relatively isotropic structure.
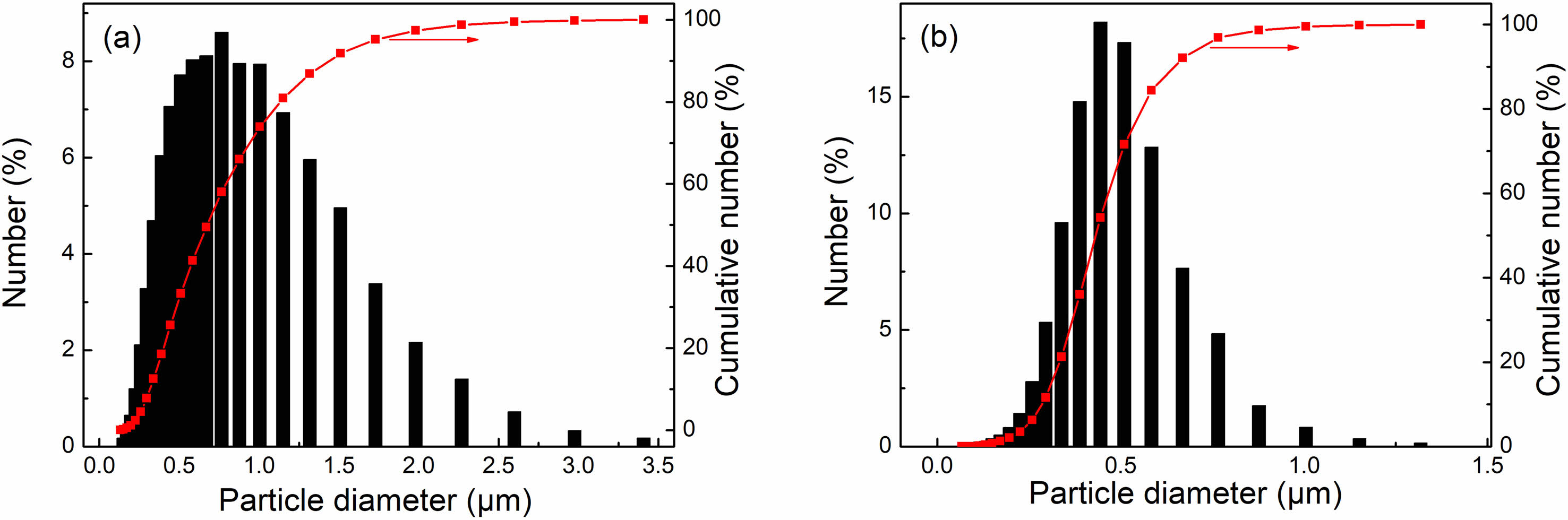
Figure 3 displays the particle size distribution and the cumulative particle size of the as-calcined powders, both undoped and doped with sulfuric acid. Both powders exhibited a single-mode distribution. Despite the small diversity of the primary particle size between undoped and doped Y2O3 powders (as shown in Fig. 2), there is a notable difference in the average agglomerate size. The undoped Y2O3 powder produced agglomerates with a maximum diameter of 3.4 µm (mean particle size D50 = 0.79 µm). Conversely, the as-doped Y2O3 powders had a narrower size distribution and a smaller D50 of 0.45 µm, indicating a more loosely agglomerated state. The particle size of doped Y2O3 powders is smaller than that of undoped ones, suggesting that adding sulfuric acid as a dopant significantly decreased the agglomeration degree of the powders during the calcination process. As demonstrated in our prior work, adding sulfate can effectively inhibit mass transfer processes associated with densification. Consequently, the addition of sulfate into Y2O3 powder during the calcination process contributed to a pronounced tendency towards particle coarsening, ultimately yielding Y2O3 powder with significantly reduced agglomeration [30].
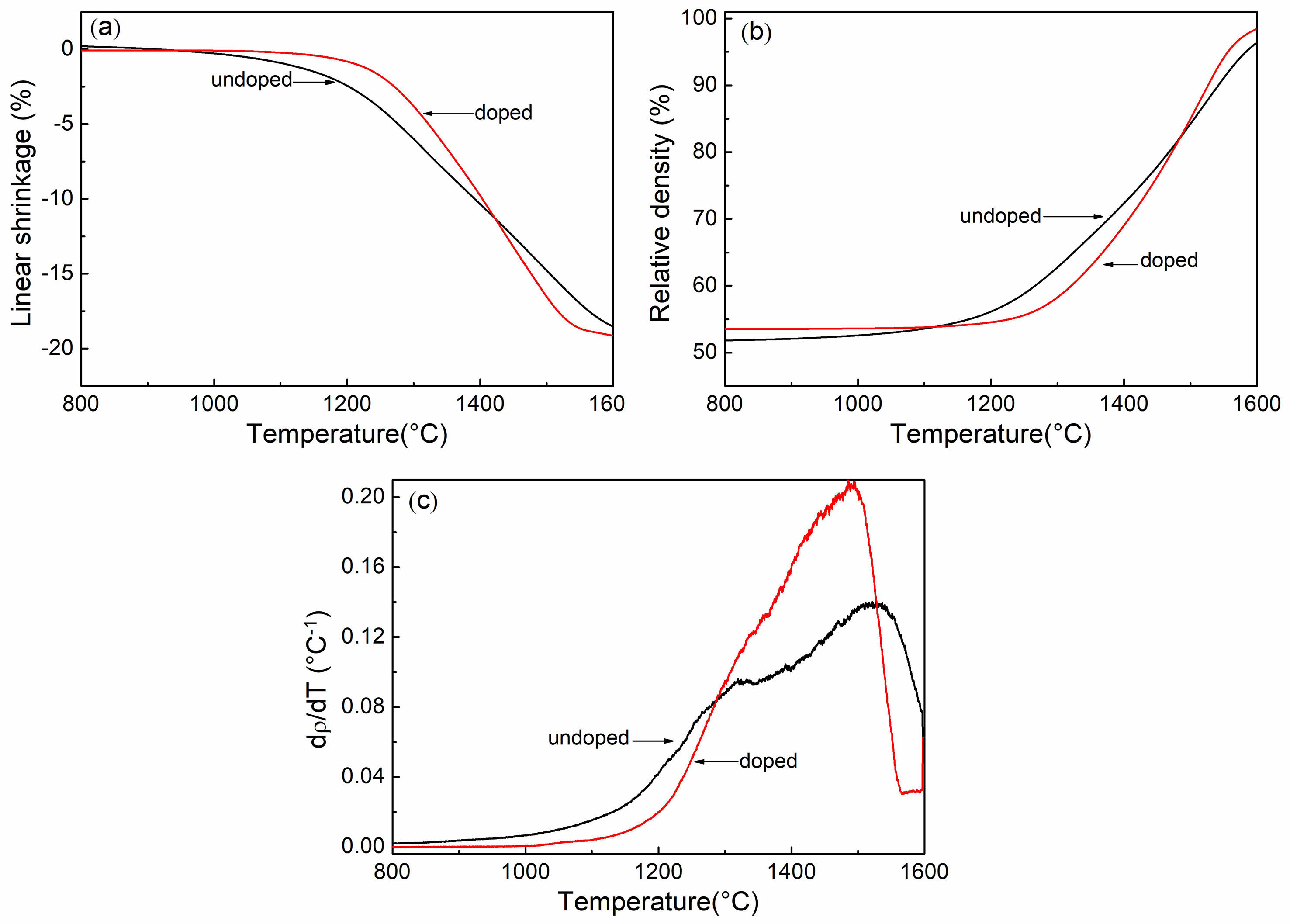
To investigate the impact of sulfuric acid on the sintering rate, the shrinkage behavior of green compacts made from both undoped and doped Y2O3 powders in a constant heating rate was examined, using a dilatometric method. Fig. 4 shows the shrinkage curves for a heating rate of 2.5 °C/min. As discernible from Fig. 4(a), the doped Y2O3 started shrinking at a slightly higher temperature (~1170 ℃) in comparison to the undoped Y2O3. Furthermore, beyond ~1425 °C, the doped Y2O3 demonstrated a higher degree of shrinkage in contrast to the undoped Y2O3. The total shrinkage was 18.4% for the undoped Y2O3 and 19.2% for the doped Y2O3. By analyzing the shrinkage curves in Fig. 4(a), the change in relative density (ρ) and densification rate (dρ/dT) with respect to sintering temperature was determined using Eq. (1) [31].

The density (ρ) of the powder compact at any given sintering temperature can be calculated using the initial green density (ρ0) and the measured linear shrinkage (∆L/L0). Here, L0 represents the initial sample length; ∆L = L-L0, L is the instantaneous sample length. As shown in Fig.4(b), At temperatures below 1250 °C, undoped Y2O3 exhibited a more rapid increase in relative density compared to doped Y2O3. The relative density of doped Y2O3 was higher than that of undoped Y2O3 at higher temperatures exceeding ~1480 ℃, and attained 98.4% of the theoretical density at 1600 ℃. Additionally, Fig. 4(c) illustrates the sintering temperature at which the densification-rate of undoped Y2O3 reached its maximum value shifted to a higher temperature of 1510 ℃ than that of doped Y2O3.
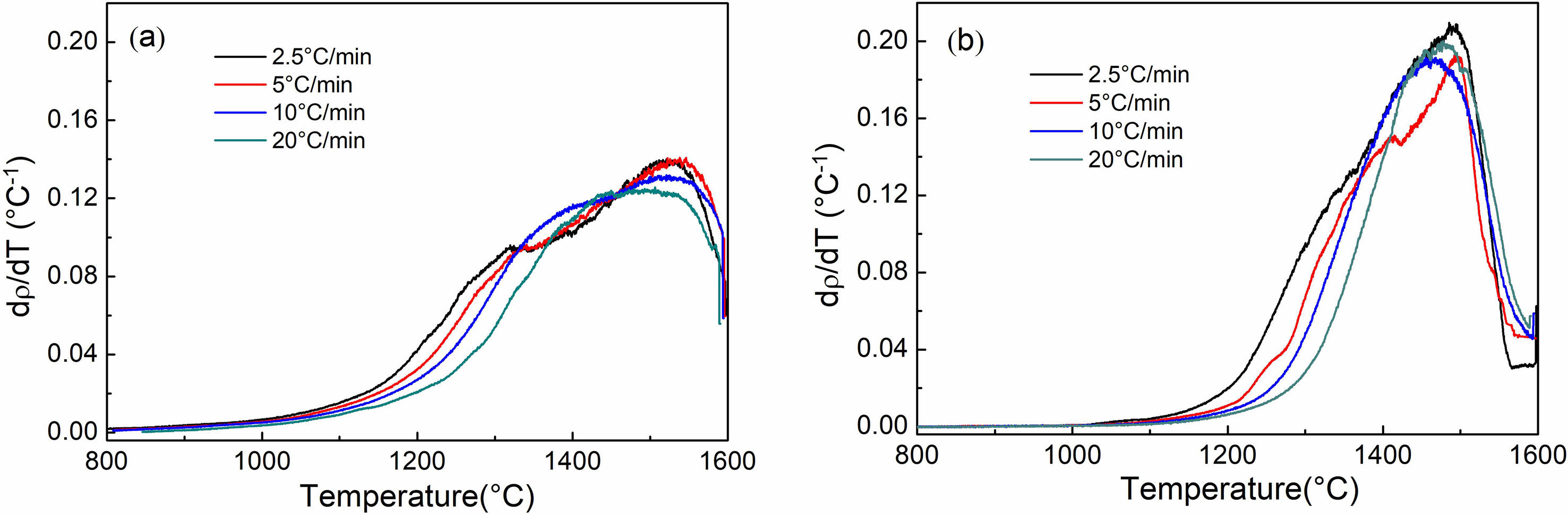
Figure 5 illustrates the densification rates of Y2O3 powders as a function of sintering temperature. These rates were observed at heating rates ranging from 2.5 to 20 ℃/min, and there was a noticeable shift in the densification rate curves toward higher temperatures, correlating with increased heating rates. At temperatures below ~1350 ℃, the doped Y2O3 exhibited a lower densification rate rather than the undoped Y2O3, regardless of the heating rate. However, beyond this temperature, the doped Y2O3 showed a higher densification rate, reaching 0.18 ℃-1 at approximately 1460 ℃, employing a heating rate of 10 ℃/min.
Wang and Raj introduced the sintering-rate equation (Eq. 2) [33, 34], which has been employed to quantitatively evaluate shrinkage and shrinkage rate data, employing the constant rate of heating (CRH) method [40].
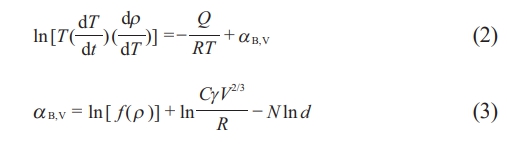
In this study, the variables used are as follows: ρ represents the density obtained from Eq. (1), Q represents the activation energy, and T represents the absolute temperature. R, C, and V are the gas constant, a constant, and the molar volume, respectively. The function f(ρ) is only dependent on density. In these models, d and N symbolize the grain size and its power law exponent, respectively. Equation (2) is applicable for fractional shrinkages below 4%, during which the initial sintering stage occurs without grain growth. A fractional shrinkage of less than 4% corresponds to a relative density range from 54% to 58% for the green compacts investigated in this study.
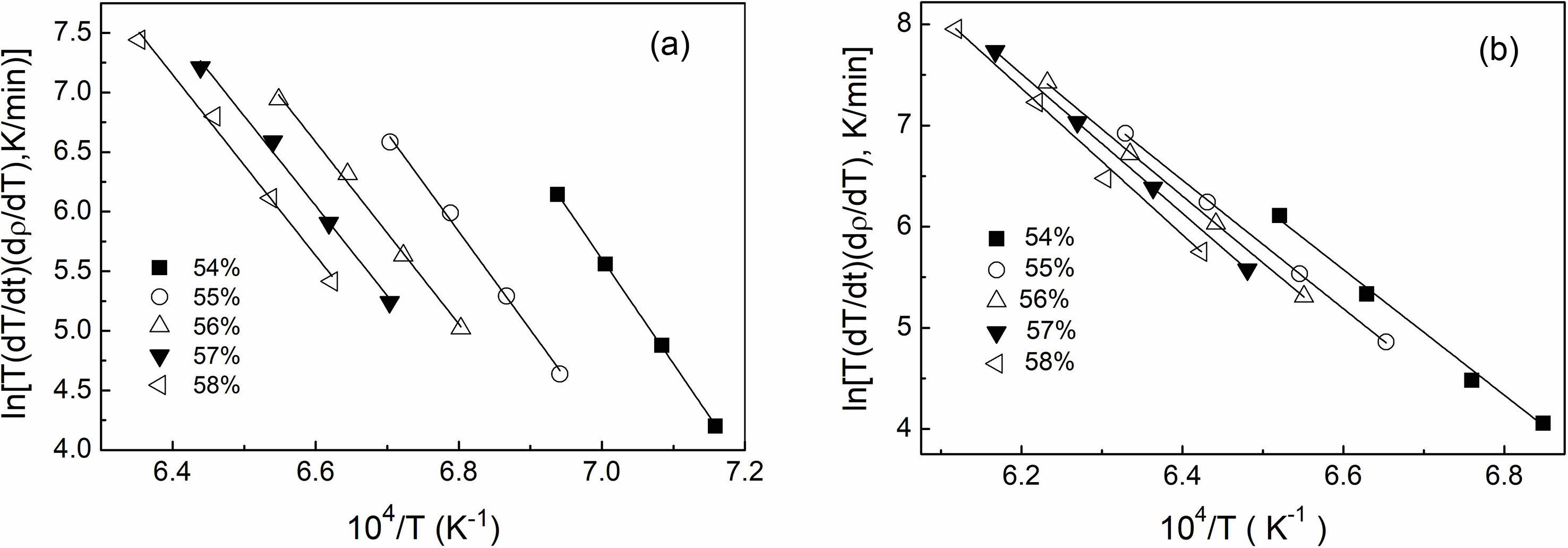
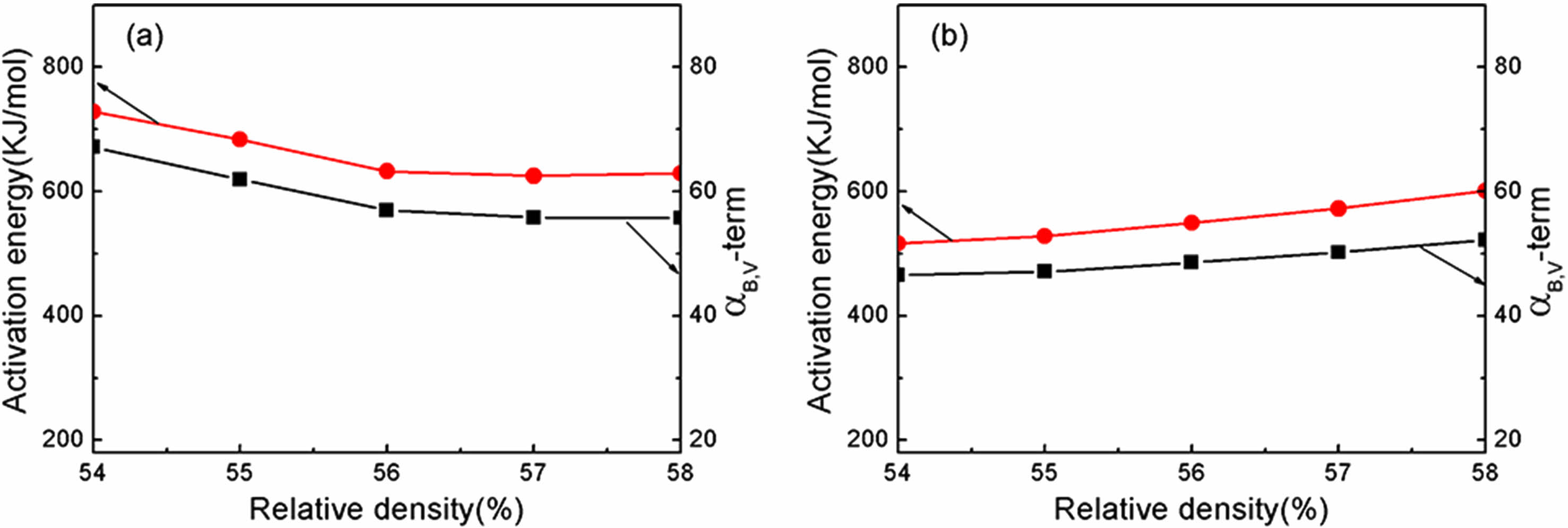
Based on the results of the densification rate of undoped and doped samples at various heating rates provided in Fig. 5, the activation energy (Q) value at each relative density can be estimated from the slope of an Arrhenius-type plot of ln[T(dT/dt)(dρ/dT)] against 1/T, as displayed in Fig. 6. The value of αB,V term was also derived from the intercept of the straight line in the plot. Fig. 7 displays the activation energy and αB,V term for the relative density range between 54% and 58%. The undoped Y2O3 exhibited significantly larger activation energies (Q) and αB,V term compared to the doped Y2O3 at each given relative density. The average activation energies for both samples were likewise calculated within the relative density range of 54% to 58%, as shown in Table 1. The undoped Y2O3 had a Q value of 656 kJ/mol, whereas the doped one only displayed a Q value (553 kJ/mol). Wang calculated the activation energy of a Y2O3 ceramic to be 410 kJ/mol [41]. In this study, the activation energies are much higher than this value, possibly due to the discrepancy of the analysis model and the particle size of powders. The agglomeration state of powder significantly impacts the activation energy required for the initial sintering process. The initial sintering process between primary particles for the doped Y2O3 with low-agglomerated particles only necessitates a low sintering temperature and activation energy because of the short diffusion distance between these particles. In the case of hard agglomerated powder for undoped Y2O3, the sintering process within these particles facilitates the formation of dense particles with larger particle sizes, increasing the diffusion mass transfer distance and eventually requiring a higher activation energy for sintering. The present results demonstrated that the addition of sulfuric acid to Y2O3 powder reduced the average activation energy during the initial sintering stage.
To determine the diffusion mechanism of undoped and doped Y2O3 powder during the initial sintering process, the activation energies of both powders were calculated using the sintering-rate eqs. (4) and (5), as given by Young [33].
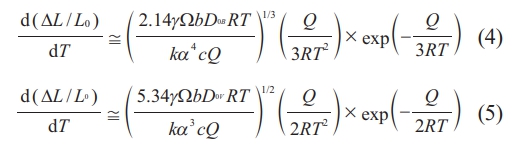
Here, eqs. (4) and (5) are employed to represent grain boundary diffusion (GBD) and volume diffusion (VD), respectively. Q is the activation energy for GBD or VD, γ denotes surface energy, Ω is atomic volume, bDB is the coefficient for GBD, bDv is the coefficient for VD, t represents time, T for absolute temperature, k for the Boltzmann constant, c for the heating rate (dT/dt), and α for the particle radius of powder.
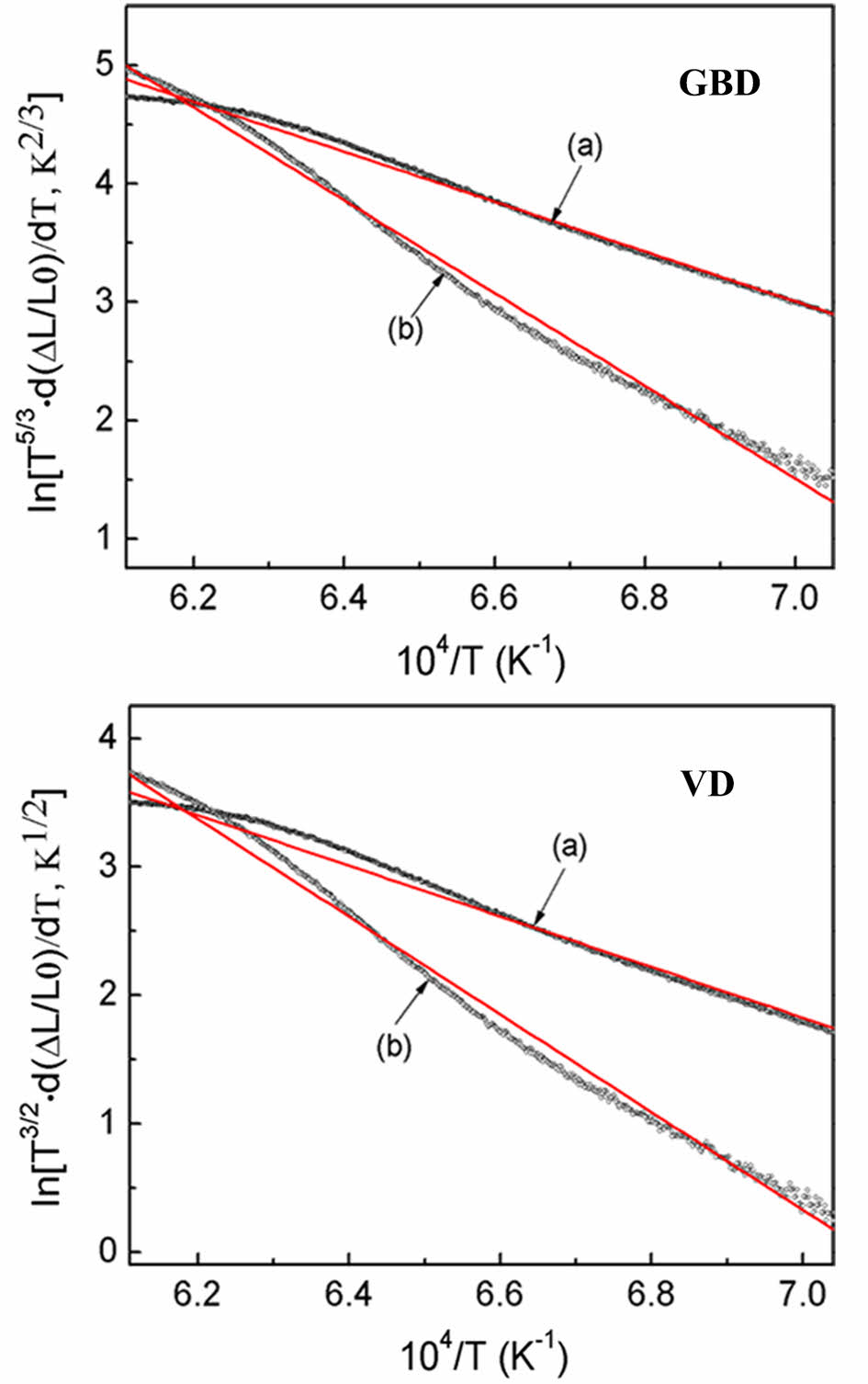
By applying eqs. (4) and (5) to the shrinkage data obtained through the CRH technique, the apparent activation energy Q/m can be calculated by analyzing the slope of the plot of ln[T 5/3 d (∆L/L0)/dT] or ln[T 3/2 d(∆L/L0)/dT] versus 1/T (GBD- or VD-type plot) [38], by assuming GBD or VD. Additionally, applying eqs. (4)-(5), the order (m) associated with the diffusion mechanism was determined during the initial sintering stage, as expressed in eq. (6).

The value of m is 3 when sintering is regulated grain boundary diffusion and 2 when it is controlled by volume diffusion.
Figure 8 displays the GBD- and VD- type plots for Y2O3 powders, employing a heating rate of 2.5 ℃/min. The slopes for doped Y2O3 exhibited a higher magnitude compared to the undoped Y2O3, indicating an increase in the apparent activation energy during the initial sintering stage, due to the introduction of sulfuric acid doping. The average apparent activation energies (Q/m) were calculated for heating rates ranging from 2.5 to 20 ℃/min, as detailed in Table 1. Eq. (6) was employed to determine the order (m), thereby elucidating that the diffusion mechanism during the initial sintering stage was investigated by evaluating the Q values and the Q/m ratio. The values of m for the (GBD) and (VD) of the undoped sample were approximately 3.3, approaching the m value of GBD, which is m = 3. This observation implies that the diffusion mechanism for undoped Y2O3 can be ascribed to GBD. The m value of the doped Y2O3, which was nearly 1.8, revealed a strong correlation with the m value characteristic of VD (m = 2), indicating that the sintering mechanism was dominated by VD. It is widely recognized that the activation energy required for GBD of oxide ceramics is lower than that of VD [38]. Therefore, it is hypothesized that the activation energy for VD in undoped Y2O3 exceeded 656 kJ/mol, based on the activation energy obtained (656 kJ/mol) for GBD in undoped Y2O3 using the present CRH shrinkage analysis. Unlike the general tendency above, the addition of sulfuric acid caused a shift in the diffusion mechanism from GBD to VD during the initial sintering stage. This change is evident in Table 1 and yielded a lower activation energy for VD (553 kJ/mol) in doped Y2O3 rather than that for GBD in undoped Y2O3. These results further suggest that the addition of sulfuric acid directly impacts the diffusion process during the initial sintering stage of nano Y2O3 powder. Interestingly, as observed in Fig. 4, compared to the undoped Y2O3, the doped Y2O3 exhibited a higher onset temperature of shrinkage, which contrasted with the activation energy of both Y2O3 powders. Our previously published research has revealed that when the calcination temperature of the powder reached 1100 °C, there were still sulfur residues in the powders. Under high-temperature conditions, the decomposition of these residues initiated a mass transfer mechanism that promoted the loose growth of the powder particles rather than densification, ultimately elevating the sintering temperature at which the doped powder began to shrink.
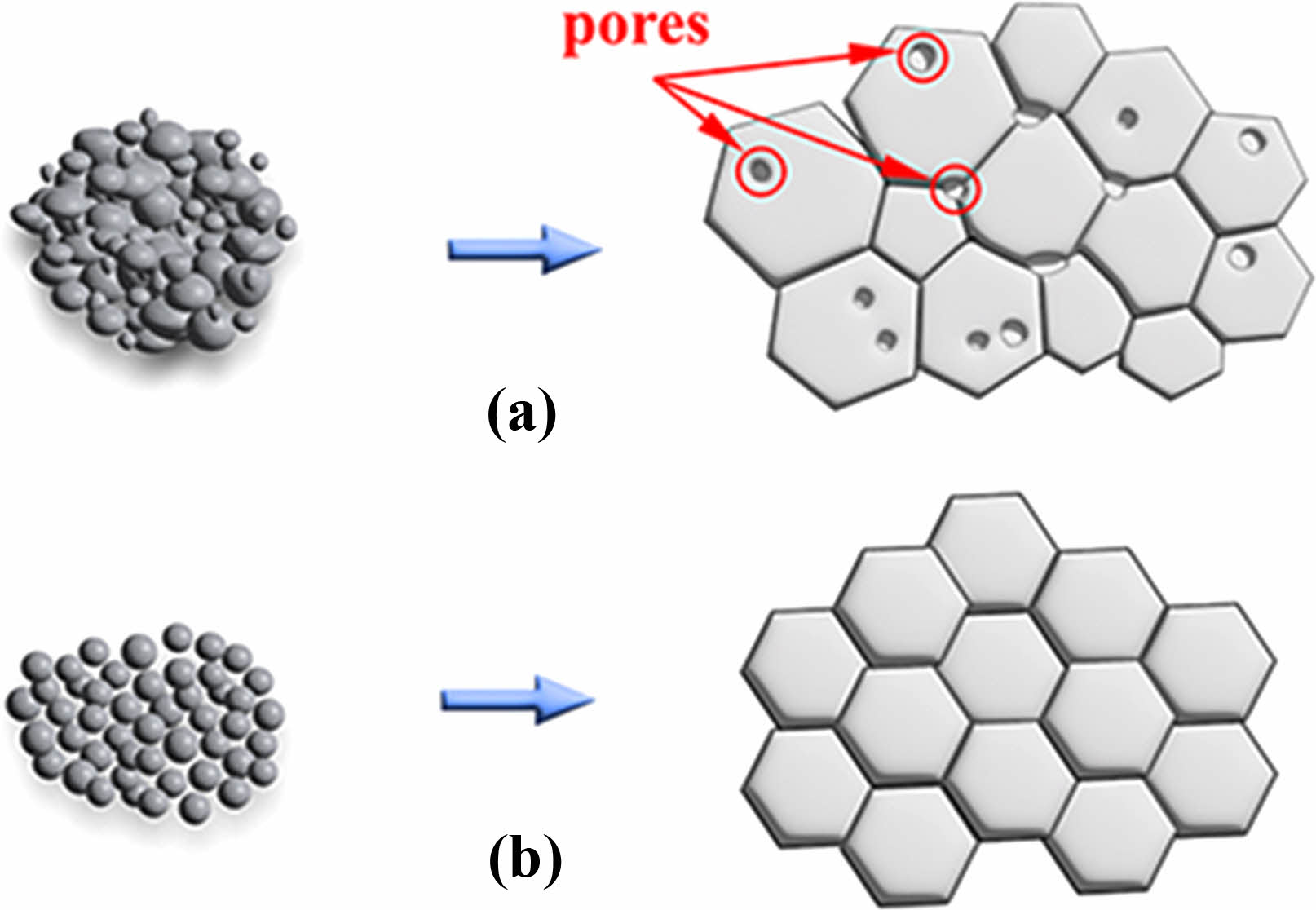
Figure 9 displays the microstructure of the fracture surfaces of Y2O3 ceramics sintered at a temperature of 1500 ℃ for 4 h in a vacuum atmosphere. The sintered ceramic produced from undoped Y2O3 powders contained a mass of pores within the grains and at the grain boundaries. This is attributed to the significantly elevated activation energy of the agglomerated Y2O3 powder. Despite employing high sintering temperatures in the final sintering stage, effectively eliminating these pores, which were wrapped in the grains, proved challenging. Moreover, the sintered ceramic obtained from doped Y2O3 powders possessed a well-defined microstructure, and only a few pores were observed (Fig. 9(b)), demonstrating their exceptional sintering activity. The enhanced sintering activity of doped Y2O3 can be attributed to its reduced activation energy during the initial sintering stage. The above results can be illustrated in Fig. 10, which is a schematic illustration of microstructures for undoped powder and doped powders and their resultant ceramics.
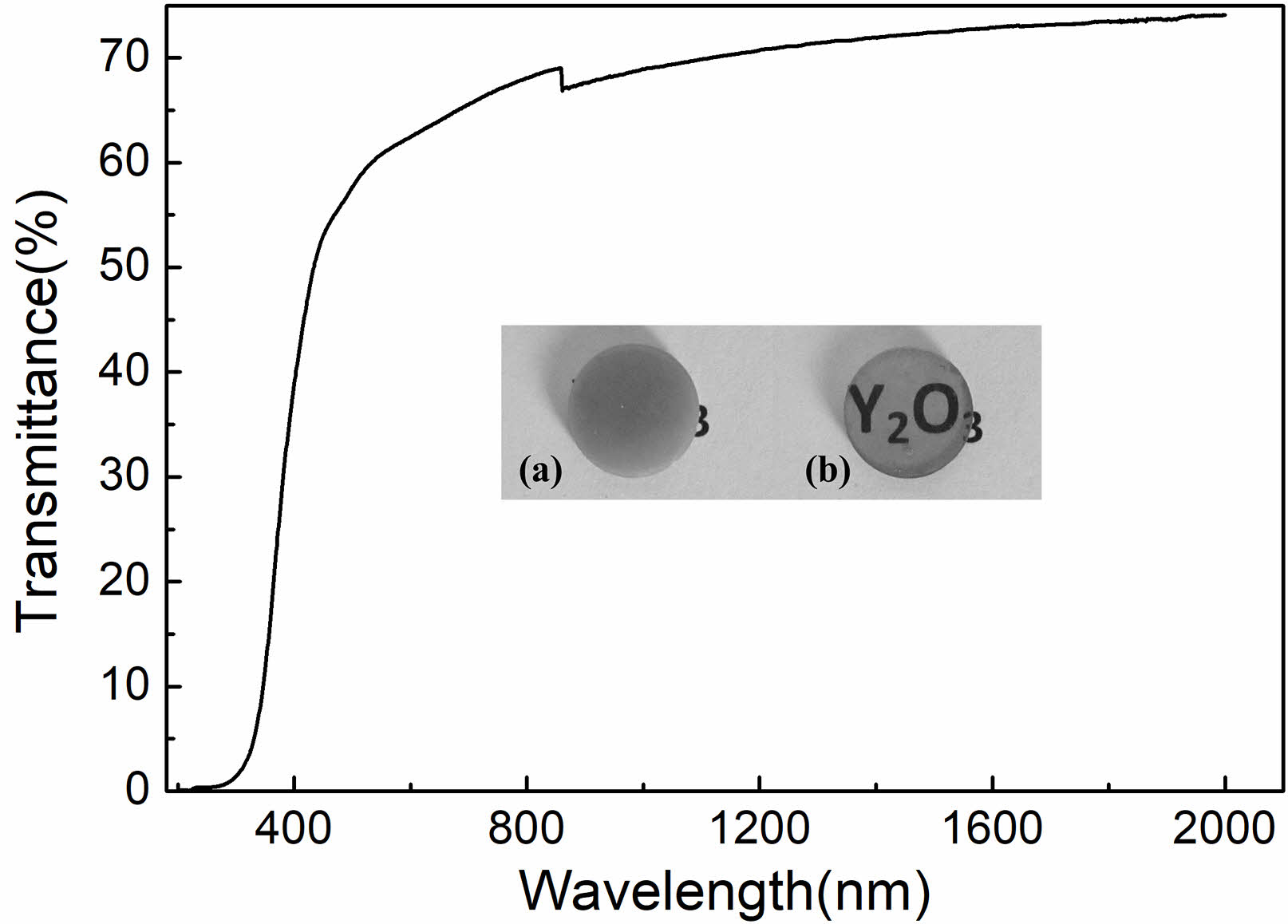
The mirror polished Y2O3 ceramic, derived from doped Y2O3 powder, had a thickness of 1.5 mm. The spectral transmittance of the ceramic was quantified across a wavelength range of 200 to 2000 nm, as shown in Fig. 11. The transmittance of the ceramic increased as the wavelength increased, reaching approximately 70% at 800 nm and 74% at 2000 nm. Conversely, the Y2O3 ceramic obtained from undoped Y2O3 powders was opaque, as shown in the inset picture in Fig. 11. The high transparency of the Y2O3 ceramic from doped powders further suggested that introducing sulfuric acid can effectively improve the sinterability of Y2O3 powders.
|
Fig. 1 XRD patterns of the calcined Y2O3 powders undoped (a) and doped with sulfuric acid (b). |

|
Fig. 2 The TEM micrographs of Y2O3 powders undoped (a) and doped with sulfuric acid (b). |
|
Fig. 3 Particle size distribution for the as-calcined powders undoped (a) and doped with sulfuric acid (b). |
|
Fig. 4 Temperature dependence of shrinkage, relative density (ρ), and densification rate (dρ/dT) of undoped powder and doped Y2O3 powder with sulfuric acid in the course of heating (2.5 ℃/min). |
|
Fig. 5 Temperature dependence of densification rate of the samples undoped (a) and doped with sulfuric acid (b) at various heating rates. |
|
Fig. 6 Arrhenius-type plots for the estimate of sintering activation energies of the samples undoped (a) and doped with sulfuric acid (b) at 54%-58% relative densities. |
|
Fig. 7 Activation energy and αB,V for diffusion of undoped and doped Y2O3 in the relative density range from 54% to 58%. |
|
Fig. 8 Grain-boundary diffusion (GBD)- and volume diffusion (VD)-type plots of the samples undoped (a) and doped with sulfuric acid (b) in the course of heating (10 ℃/min). |

|
Fig. 9 SEM micrographs of the fractured Y2O3 ceramic surfaces. undoped (a) and doped (b) Y2O3. |
|
Fig. 10 Schematic illustration of microstructures for undoped powder (a) and doped powders (b), as well as their resultant ceramics. |
|
Fig. 11 In-linear transmittance of 1.5-mm thick Y2O3 ceramic. Inset showing a photograph of polished Y2O3 ceramics derived from undoped (a) and doped (b) powders. |
|
Table 1 Apparent and activation Energy, and Order Depending on the Diffusion Mechanism. |
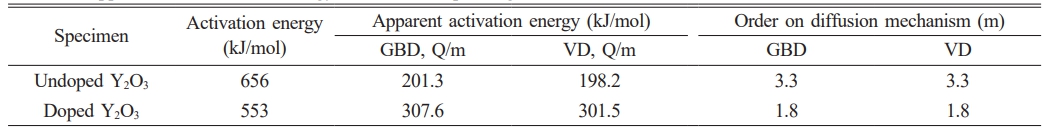
Transparent Y2O3 ceramics were prepared from nano-sized, sulfuric acid-doped Y2O3 powders using a vacuum sintering method. The doped Y2O3 powders revealed particles of finer size and less aggregates instead of the undoped Y2O3 powders. The sintering behaviors of powder compacts at the initial sintering stage were assessed using the CRH method. The addition of sulfuric acid remarkably increased the densification rate of Y2O3 powders, especially above ~1350 ℃, yielding a decline in the average activation energy (Q value) from 656 kJ/mol for the undoped Y2O3 to 553 kJ/mol for the doped Y2O3 in the initial sintering stage. Besides, it caused a shift in the diffusion mechanism from GBD to VD. Upon vacuum sintering at 1500 ℃, the ceramic for doped powders exhibited increased density and reduced residual pores compared to undoped one. In the case of the doped powder with sulfuric acid, a dense, transparent Y2O3 ceramic, with 74% transparency at 2000 nm wavelength, was achieved under vacuum sintering at 1700 ℃ for 5 h, unlike the opaque ceramic produced from undoped Y2O3.
This work was supported by the National Natural Science Foundation of China (Grant No. 51802136), Foundation of Liaoning Educational Committee (Grant No. L2020043).
- 1. A. Ratsimba, A. Zerrouki, N. Tessier-Doyen, B. Nait-Ali, D. André, P. Duport, A. Neveu, N. Tripathi, F. Francqu, and G. Delaizir, Ceram. Int. 47[6] (2021) 7465-7474.
-

- 2. D.S. Lee, H. Kong, and S.J. Lee, J. Ceram. Process. Res. 23[2] (2022) 137-144.
-
- 3. H.M. Oh, Y.J. Park, H.N. Kim, J.W. Ko, and H.K. Lee, Ceram. Int. 47[4] (2021) 4681-4687.
-
- 4. C.W. Park, J.H. Lee, S.H. Kang, J.H. Park, H.M. Kim, H.S. Kang, H.A. Lee, J.H. Lee, and K.B. Shim, J. Ceram. Process. Res. 18[3] (2017) 183-187.
-
- 5. D. Permin, O. Postnikova, S. Balabanov, A. Belyaev, V. Koshkin, O. Timofeev, and J. Li, Materials. 16[1] (2023) 260.
-
- 6. Y.G. Yang, J.Y. Kwak, H. Kong, and S.J. Lee, J. Ceram. Process. Res. 21[4] (2020) 450-455.
-
- 7. L. Gan, Y.J. Park, M.J. Park, H. Kim, J.M. Kim, J.W. Ko, and J.W. Lee, J. Am. Ceram. Soc. 98[7] (2015) 2002-2004.
-
- 8. L.L. Zhu, Y.J. Park, L. Gan, S.I. Go, H.N. Kim, J.M. Kim, and J.W. Ko, J. Mater. Sci. Mater. Electron. 28 (2017) 7854-7861.
-
- 9. H.B. Zhang, B.N. Kim, K. Morita, H. Yoshida, K. Hiraga, and Y. Sakka, J. Am. Ceram. Soc. 94[10] (2011) 3206-3210.
-
- 10. R. Chaim, A. Shlayer, and C. Estournes, J. Eur. Ceram. Soc. 29[1] (2009) 91-98.
-
- 11. H.M. Oh, H.N. Kim, Y.J. Park, J.W. Ko, H.K. Lee, S.J. Hwang, and T.J. Yu, J. Eur. Ceram. Soc. 41[7] (2021) 4419-4423.
-
- 12. V.V. Osipov, A.N. Orlov, R.N. Maksimov, V.V. Lisenkov, and V.V. Platonov, Phys. Status Solidi C. 10[6] (2013) 914-917.
-
- 13. C.W. Park, J.H. Park, H.S. Kang, H.A. Lee, J.H. Lee, J.H. In, and K.B. Shim, J. Ceram. Process. Res. 19[5] (2018) 383-387.
-
- 14. C. Greskovich and J.P. Chernoch, J. Appl. Phys. 44 (1973) 4599-4606.
-
- 15. K. Majima, N. Niimi, M. Watanabe, S. Katsuyama, and H. Nagai, J. Alloys Compd. 193[1-2] (1993) 280-282.
-
- 16. L. Gan, Y.J. Park, H. Kim, J.M. Kim, J.W. Ko, and J.W. Lee, Ceram. Int. 41[8] (2015) 9622-9627.
-
- 17. Z.C. Fu, N. Wu, H.B. Long, J.M. Wang, J. Zhang, Z.X. Hou, X.D. Li, and X.D. Sun, Coatings. 12[8] (2022) 1077.
-
- 18. H. Eilers, J. Eur. Ceram. Soc. 27[16] (2007) 4711-4717.
-
- 19. X.R. Zhang, W.Z. Lu, G.F. Fan, and X.H. Wang, Adv. Powder Technol. 27[1] (2016) 295-298.
-
- 20. R.P. Yavetskiy, D.Y. Kosyanov, V.N. Baumer, A.G. Doroshenko, A.I. Fedorov, N.A. Matveevskaya, A.V. Tolmachev, and O.M. Vovk, J. Rare Earths. 32[4] (2014) 320-325.
-
- 21. T. Onodera, T. Ikegami, Y. Yajima, M. Kawamura, M. Sakai, and Y. Moriyoshi, J. Ceram. Soc. Jpn. 111 (2003) 664-668.
-
- 22. H.M. Qin, H. Liu, Y.H. Sang, Y.H. Lv, X.L. Zhang, and Y.Y. Zhang, Cryst Eng Comm. 14 (2012) 1783-1789.
-
- 23. H.M. Qin, X.H. Zhang, H. Liu, Y.H. Sang, and J.Y. Wang, Cryst Eng Comm. 15 (2013) 5076-5081.
-
- 24. Y. Liu, X. Qin, H. Xin, and C. Song, J. Eur. Ceram. Soc. 33[13-14] (2013) 2625-2631.
-
- 25. L. Wen, X.D. Sun, Z.M. Xiu, S.W. Chen, and C.T. Tsai, J. Eur. Ceram. Soc. 24[9] (2004) 2681-2688.
-
- 26. Z.G. Huang, X.D. Sun, Z.M. Xiu, S.W. Chen, and C.T. Tsai, Mater. Lett. 58[15] (2004) 2137-2142.
-
- 27. J. Li, W.B. Liu, B.X. Jiang, J. Zhou, W.X. Zhang, L. Wang, Y. Shen, Y. Pan, and J. Guo, J. Alloys. Compd. 515 (2012) 49-56.
-
- 28. S.S. Li, B.L. Liu, J. Li, X.W. Zhu, W.B. Liu, Y.B. Pan, and J.K. Guo, J. Alloys. Compd. 678 (2016) 258-266.
-
- 29. L. Zhang, Z. Li, F.Z. Zhen, L.X. Wang, Q.T. Zhang, R. Sun, F.A. Selim, C.P. Wong, and H. Chen, J. Mater. Sci. 52 (2017) 8556-8567.
-
- 30. J. He, X.D. Li, S.H. Liu, Q. Zhu, J.G. Li, and X.D. Sun, J. Eur. Ceram. Soc. 35[8] (2015) 2369-2377.
-
- 31. W. Cao, X. Mao, Y. Yuan, L. Li, L.B. Zhao, and J. Li, J. Eur. Ceram. Soc. 37[13] (2013) 4005-4013.
-
- 32. W.M. Zeng, L. Gao, L.H. Gui, and J.K. Guo, Ceram. Int. 25[8] (1999) 723-726.
-
- 33. W.S. Young and I.B. Cutler, J. Am. Ceram. Soc. 53[12] (1970) 659-663.
-
- 34. J. Wang and R. Raj, J. Am. Ceram. Soc. 74[8] (1991) 1959-1963.
-
- 35. J. Wang and R. Raj, J. Am. Ceram. Soc. 73[5] (1990) 1172-1175.
-
- 36. K. Matsui, A. Matsumoto, M. Uehara, N. Enomoto, and J. Hojo, J. Am. Ceram. Soc. 90[1] (2007) 44-49.
-
- 37. L.A. Pérez-Maqueda, J.M. Criado, and C. Rea, J. Am. Ceram. Soc. 85[4] (2002) 763-768.
-
- 38. K. Matsui, N. Ohmichi, and M. Ohgai, J. Am. Ceram. Soc. 88[12] (2005) 3346-3352.
-
- 39. K. Matsui, K. Tanaka, T. Yamakawa, M. Uehara, N. Enomoto, and J. Hojo, J. Am. Ceram. Soc. 90[2] (2007) 443-447.
-
- 40. K. Matsui, J. Am. Ceram. Soc. 91[8] (2008) 2534-2539.
-
- 41. X.H. Wang, P.L. Chen, and I.W. Chen, J. Am. Ceram. Soc. 89[2] (2006) 431-437.
-
 This Article
This Article
-
2024; 25(3): 404-412
Published on Jun 30, 2024
- 10.36410/jcpr.2024.25.3.404
- Received on Mar 12, 2024
- Revised on May 18, 2024
- Accepted on May 22, 2024
Services
- Abstract
introduction
experimental procedure
results and discussion
conclusions
- Acknowledgements
- References
- Full Text PDF
Shared
Correspondence to
- Jiao He
-
School of Mechanical Engineering, Liaoning Petrochemical University, Fushun, 113001, P.R. China
Tel : +86-24-56865042 Fax: +86-24-56865042 E-mail: hejiao@lnpu.edu.cn - E-mail: hejiao@lnpu.edu.cn
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr