





- The influence of the nature and functionalization of the nanoparticuled fillers on the performances of the dental nanocomposite
Ammar Ali Husseinaa,*, Mohammed Ali Mutarb and Anton Ficaic
aScience and Engineering of Oxide Materials and Nanomaterials, Faculty of Chemical Engineering and Biotechnology, University POLITEHNICA of Bucharest, Gh. Polizu 1-7, Bucharest, Romania
Department of Science and Engineering of Oxide Materials and Nanomaterials, Faculty of Chemical Engineering and Biotechnologies, University Politehnica of Bucharest, 1-7 Gh. Polizu Street, 011061 Bucharest, Romania
Ministry of Education, The General Directorate of Educational in Najaf Al-Ashraf, Najaf, Iraq
bDepartment of Chemistry, College of Sciences, University of AL Qadisiyah, Iraq
cAcademy of Romanian Scientists, 3Ilfov Street, 050044 Bucharest, RomaniaThis article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
The current work focuses on the modification of dental nanocomposites using nanosized fillers. To determine the impact on the wear resistance, flexural strength, compressive strength, and hardness qualities of the dental nanocomposites, three types of nanoparticles—silica (SiO2), zircon (ZrO2), and hydroxyapatite (H.A.: Ca5(PO4)3OH)—are combined with a standard matrix of dental resin. This study’s objective is to create dental nanocomposites using the nanoparticles above as fillers in Bis-GMA with unsaturated monomers, urethanedimethacrylate (UDMA), methylmethacrylate (MMA), methacrylic acid (MAA), and bisphenol A dimethacrylate in the presence of 1,6-hexanediol methacrylate (HDODA) as a crosslinking agent. To start the copolymerization of matrix resins, camphor quinone (C.Q.) of 0.5 wt.% and 2-(dimethyl amino) ethyl methacrylate (DMAEMA) of 0.5 wt.% are utilized as photoinitiation systems. These nanocomposites have the potential for posterior restorative applications. Treatment of the Nano (SiO2, ZrO2, Hydroxyapitate (HA)) particles was carried out with a silane coupling agent, 3-(methacryloyloxy)propyltrimethoxysilane (MPTMS), to improve bonding between the Nano particles and resin matrix, and reduce agglomeration of the Nano SiO2,ZrO2, Hydroxyapitate (HA)). Characterization of products was carried out using scanning electron microscopy (SEM), and Fourier transform infrared spectroscopy (FTIR). SEM images showed the adhesion between the resin matrix and the Nano (SiO2, ZrO2, Hydroxyapitate (HA)), and particle size distribution in addition to the particle agglomeration that is related to the treated Nanofillers in Nanocomposites. FTIR was used to show the qualitative composition of untreated Nanofillers and treated Nanofillers. The three types of nanoparticles (SiO2, ZrO2, and H.A. with an average size between 10 and 30 nm) were further functionalized with 3-(methacryloyloxy)propyltrimethoxysilane (MPTMS), a silane coupling agent, to improve compatibility between the phases. Using scanning electron microscopy (SEM) and Fourier transform infrared spectroscopy, the compatibility between the phases and the morphology was determined (FTIR). The adhesion between the resin matrix and the particle, whether homogeneous or heterogeneous nanocomposites, are formed. SEM saw the effect of salinization. FTIR was employed to communicate qualitative information regarding the impact of salinization. flexural strength, and compressive strength, of nanocomposites made with untreated nanoparticles or nanoparticles treated with 2.5 wt.% MPTMS were significantly greater than those made with nanoparticles treated with 1.5 or 3.5 wt.% MPTMS. The nanocomposites containing 10 wt.% SiO2 had more flexural strength, compressive strength than those containing other nanofillers (ZrO2 and hydroxyapatite (H.A.). Surprisingly, this development was observed at a significantly lower nanosized filler concentration. Utilizing the TGA and DSC methodologies, physicochemical parameters such as solubility (S.L.), water sorption (W.S.), and volumetric shrinkage (VS) were also investigated. Additionally, the thermal stability of the entire dental nanocomposites is evaluated.
Keywords: Nanoparticles fillers, Coupling agent MPTMS, flexural strength ,Compressive strength , Light curing.
The primary objective of dentistry and dental technology is to protect or enhance the living quality of dental patients. This purpose can be attained by avoiding sickness, eliminating discomfort, enhancing mastication, enhancing speech, and enhancing beauty. Replacing or restructuring the tooth structure is necessary to accomplish several of these goals. For decades, the most significant obstacles have been the development of biocompatible, durable, direct-filling, and indirect-processed prosthetics that can withstand the adverse circumstances of the oral environment [1]. Dental materials can be used for preventive restorations. The four categories of dental materials are metals, ceramics, polymers, and composites. Despite recent improvements in these materials’ properties, no one of these materials is long-lasting. The ideal characterization of a hypothetical restorative material would account for its (1) biocompatibility, (2) ability to bond steadily to the structure of tooth or bone, (3) “ability to mimic the natural appearance of tooth enamel, dentin, and other tissues, (4) ability to determine properties similar to those of tooth enamel, dentin, and other tissues, and (5) ability to initiate tissue repair or regeneration of damaged tissues or the missing ones” [1]. The evolution of resin-based composites has streamlined the essential elements for cosmetic and practical repairs. Bisphenol, A glycidyl methacrylate Bis GMA-based resin, has shown promise as a dental composite material since its introduction [2]. Since its introduction, various dental resins have been created to address shrinkage, wear resistance, and poor strength. Furthermore, the size of the inorganic filler is a crucial component that may have a substantial impact on those properties. Because of their low durability and harsh textures, posterior restorations could not use the first generation of composites [2]. Such was because the fillers’ particle size manipulated in this composite was large. During the 1970s, micro-filled composites consisting of silica with a size particle almost 40 nm were improved to develop wear resistance and provide a lustrous surface. The hybrid composites’ mechanical properties have been enhanced by adding glass particles (of various sizes) as fillers, such as quartz SiO2, aluminosilicate (Al2SiO5), and silica glasses [3]. Certain developments are there that are continuing to deliver proper dental materials for clinical achievement [4]. Continuous investigations to drop the fillers’ size to develop properties have lately resulted in dental composites expansion based on nanotechnology. The manufacturing of functional materials is based on nanotechnology engineering, using various physical and chemical methods that allow the development of particles between 0.1 and 100 nm. Nanohybrid materials can be obtained, but new approaches must be compared to traditional materials obtained with larger filler. Using either method can improve the quality of the composite material, although it may result in the premature loss of bigger particles and/or gloss [4]. The aesthetic qualities of restorative nanocomposite materials include a high polishing ability and a low degree of polymerization shrinkage. Aesthetic enhancements, such as increased translucency and gloss and optimized strength and wear resistance, are driving developments in restorative nanocomposite materials. These materials currently deliver clinicians a reliable option for anterior and posterior restorations [5]. The result of the global recognition related to the poor aesthetic value as well as the effects of amalgam that is potentially toxic is the rise of replacing amalgams with dental composites. This is done even though amalgams often exhibit superior wear characteristics than composites in the back of the mouth [6]. Composites have recently become a popular alternative to amalgams for dental restorations [7]. Although composite was originally and primarily used for small restorations, its application has grown to include indirect replacement for major cusp-bearing restorations, crowns, and bridges. Even though dental nanocomposites are increasingly used as restorations in large stress-bearing defects and as a replacement for amalgam inlay, there are still several important concerns regarding their use, including wear, flexibility, and flexural strength [8]. The mechanical properties of dental nanocomposites are mostly studied in simulated oral conditions. Some complex processes are associated with restorative nanocomposite materials, such as masticatory stress in the presence of saliva, a chemical environment that can aid in the degradation of the restoration material, and some physical factors, such as the large temperature fluctuations associated with the consumption of cold/hot foods. Considering all of these factors and obstacles, this manuscript aims to investigate the enhancements of dental nanocomposites based on different resins and appropriate nanofillers (SiO2, ZrO2, HA) with an average size of 10-30 nm, functionalized using MPTMS as a coupling agent.
Materials
Methylmethacrylate (M.A.) (MERCK), Methacrylic Acid (MAA), (HiMedia Leading Biosciences Co.), 2,2-bis[4(2-hydroxy-3methacryloylpropyloxy)phenyl] propane (Bis-GMA), Urethanedimethacrylate (UDMA), (USA), 1,6-hexanediol methacrylate (HDODA)) Bisphenol
A dimethacrylate, 3-(methacryloyloxy) propyltrimethoxysilane (MPTMS), (Aldrich), Zinc Oxide Nanoparticles (ZnO) (GCC), “Zirconium Oxide Nanoparticles ZrO2 Skyspring Nanomaterial, Silicon Dioxide Nanoparticles (SiO2) Skyspring Nanomaterial, Nanohydroxy apatite (H.A.) (Skyspring Nanomaterial), Camphorquinone, 2-(Diethyl amino)ethyl acrylate (ALDRIH).”
Apparatus
“Electronic Sensitive Balance Germany, Sartorius, Bl210s FEI Nova Nano SEM 230, BRUKER, Germany, FTIR TENSOR 27, Fourier transform infrared spectrometer, Differential thermal analysis. In the College of Education for Pure Sciences Ibn-AL Haitham Central Service Laboratory at the University of Baghdad, DSC was performed using SETARAM France’s DSC 131 Evo instrumentation. Thermogravimetric studies of TGA were performed on a T.G. analyzer Model pL T.G. at Iran Polymer, Petrochemical Institute in England at a heating rate of 10 oC. A machine that can do tensile testing at room temperature with a load cell of 20 kN and a cross-head speed of about 200 mm/min is used to analyze mechanical properties. The sample dimensions complied with ASTM D-412 specifications”.
Experimental design
This investigation is based on three factors. The first part relates to the material design, based on a comparative study employing the ZrO2, SiO2, and HA-based nanocomposites for dental applications. The second section is experimental and gives data on the created ZrO2, SiO2, and H.A. nanocomposites as well as regulator composite materials in terms of wear, flexural strength compressive, wear resistance, hardness, and compression. The third part relates to experimental characterization analysis and is meant to generate suggestions for the general presentation of the estimated dental nanocomposite ingredients.
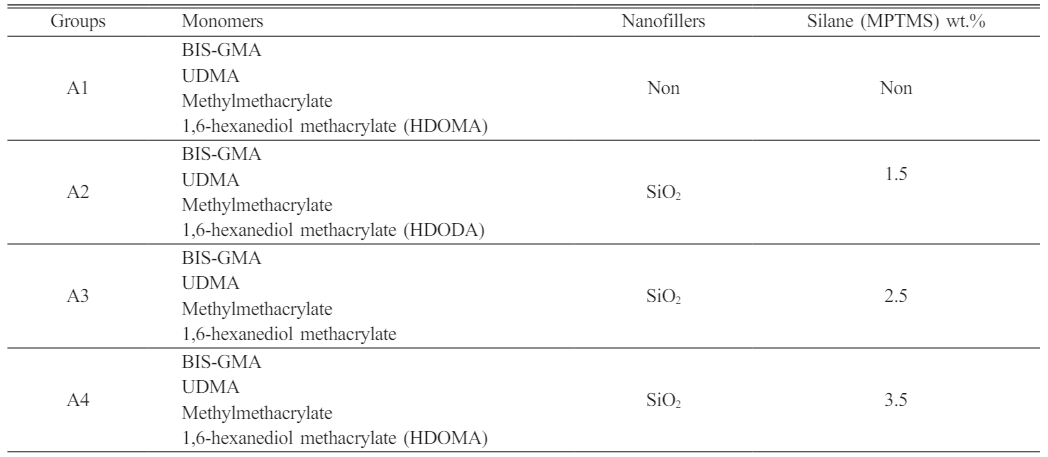
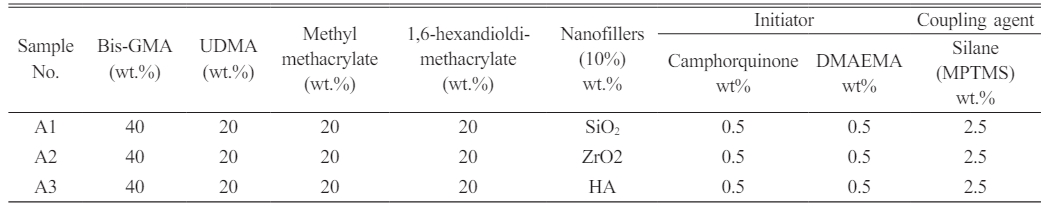
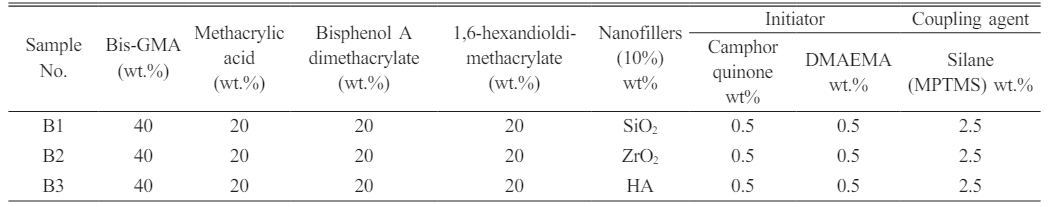
Dental nanocomposite materials were created based on the resin groups employed (denoted with DNC A and DNC B). The monomer matrix was homogenized with 10 percent by weight of various nano-fillers. One group is treated with MPTMS-treated nanoparticles at three unique weight per cent concentrations of 1.5, 2.5, and 3.5. The silanization agent was employed to evaluate the adhesion of the resin to the filler surfaces (Table 1). Table 2 Table 3
Surface treatment of Nano-SiO2
The surface treatment of Nano-SiO2 is accomplished by dissolving MPTMS in xylene, followed by the addition of Nano-SiO2 to achieve three distinct concentrations of MPTMS (1.5, 2.5, and 3.5 wt.%) in separate tests. The salinization was done in acidic conditions using acetic acid, with a pH of 3.3 and a value of 3.3 [9]. This mixture is sonicated for 10 minutes in place of mechanical stirring, then centrifuged at 10000 rpm for 10 minutes to separate the silanized nano-SiO2. Before and after centrifugation, the suspensions were rinsed with ethanol and then dried for 12 hours at 80 °C in an oven. FTIR spectroscopy was applied to both treated and untreated Nano-SiO2 to determine the chemical changes that occur on the surface.
Preparation of dental nanocomposites
The corresponding Nanocomposites’ series, called A and B (depended upon the particle sizes of ZrO2, SiO2, and H.A. (fillers). Every series contains three groups, made by mixing the monomers resin matrix and 10 wt.% of Nano-ZrO2, SiO2, and H.A. (fillers) [10]. The subsequent chemicals are manipulated: 0.5 weight per cent C.Q. acts as an initiator, while 0.5 weight per cent DMAEMA acts as an accelerator. To the manufacturer’s instructions for the control, the nanocomposite was homogenized using a magnetic stirrer in a glass container before being further irradiated for 60 seconds. The specimens are 2 mm, 4 mm, and 25 mm.
Six specimens, separated into two groups (A and B), were utilized in this investigation. Each group was exposed to a manufacturing procedure based on the composite resins used to create the specimen, including mixing Nanofillers and the monomer matrix.
The Process of Curing
The dental nanocomposite is cured using a combination of heating and light irradiation. The lamp emits light between 400 and 580 nanometers and is responsible for initiating the curing of the monomeric component. The Lumamat® 100 was utilized to both cure and temper the nanocomposites. The commercially available light sources modified for photocuring emit visible radiation in the 480 nm blue spectrum range. To meet the requirements for dental applications, it is necessary to employ safe and effective photo-starting systems. They are composed chiefly of camphor quinone (C.Q.) and many amines, such as 2-(Diethyl amino)ethyl acrylate (DMAEMA).
Volumetric Shrinkage Measurement (VS)
Following the Archimedes principle, the VS of dental resins is determined by comparing differences in density before and after photopolymerization. The evaluation is conducted using commercially available Density Determination Kits and a Mettler ToledoX analytical balance by ISO17304:2013 (E). A glass dish is utilized to determine the density of the unpolymerized material. Initially, the density of the glass dish (gd) was estimated by weighing the dish in both water and air and applying the equation.

The Water signifies mgd1, and mgd2 represents the density of glass made of the dish in the water.
The density of the glass dish has been determined in triplicate by the mean value ρgd,m when dispensing a certain amount of the sample that is un-polymerized in the glass dish and then weighing the total mass associated with the dish made of glass word well as the sample in water and air too, density a related the density of the sample that is unpolymerized ρup was estimated using eq. (2)
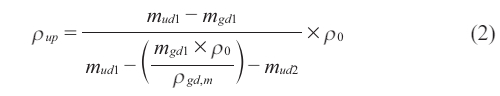
Where mgd1 denotes the weight of the glass dish in air, mud1 the weight of the glass dish and the unpolymerized sample in air, and mud2 the weight of the glass dish and the unpolymerized sample in water; Glass dish density is shown as (gd,m), while water density is shown as 0.
To follow the measurement process, the glass dish was cleaned thoroughly. There is a repetition of the measurement process on 4 extra samples to acquire a mean value connected with density concerning the un-polymerized ρup,m.
As can be seen in the D.C. measurements (the 60s to 1 part up to the time when the total sample was irradiated), the resins were first poured into a steel mould with dimensions of (2 mm × 2 mm × 25 mm), and then light-cured using a specific light dental source. The polymerized sample’s mass (size) was weighed in water, and in air, polymerized sample’s density ρps was assessed according to eq. (3)

“Where ρ0 represents the density of water, mps1 represents the mass of the polymerized sample, in the air; mps2 represents the mass of the polymerized sample, in the water. The mean value of the density of the polymerized sample, ρps,m was determined based on the 5 individuals ρps values”.
Finally, the volumetric shrinkage sample was assessed according to Eq. (4):

Measurement of Water Sorption (W.S.)
Between two translucent Mylar sheets, the resins were poured into a 1.00 mm tall, 15 mm inner diameter, cylinder-shaped steel mold. The upper surface was covered with a 1 mm thick glass slide. The samples were then exposed to light for 60 seconds using a dental light source equal to what was observed in D.C. tests. Currently, five specimens of each dental resin system are being fabricated. The initial weight (M1) connected to each specimen was determined using an electronic balance (MettlerA30, Mettler Instrument Co., High Stone, NJ, USA) with a weighing precision of (0.1 mg). The specimens were then placed in 30 millilitres of distilled water and stored at 37 degrees Celsius. The fish were then withdrawn at predetermined intervals, patted dry to eliminate extra water, reweighed, and reintroduced to the water. At thirty days of immersion, no substantial variations in mass have been seen except for M2’s equilibrium mass. Specimens were dried at a temperature of 60 oC until their mass remained constant, after which the result was recorded as M-3. Equations were utilized to evaluate W.S. (5):
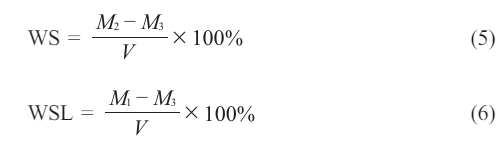
Scanning Electron Microscopy (SEM)
The cracked surfaces of the cured nanocomposite materials were studied morphologically using a Hitachi TM3000 scanning electron microscope in Tokyo, Japan. Before analysis, gold is sputter deposited onto sample surfaces.
Physico-mechanical properties of the dental nanocomposites
Flexural characteristics
The flexural strength (σ) and the flexural modulus € are indicated and determined according to the ISO178 tests of 3-point bending by using a universal testing machine LARYEE Co, China [11]. Rectangular polymer samples of 80 mm × 10 mm × 4 mm (length × width × thickness) were cut out of the molds and used for the experiments. E as well as σ were determined according to Eqs. (7) and (8):
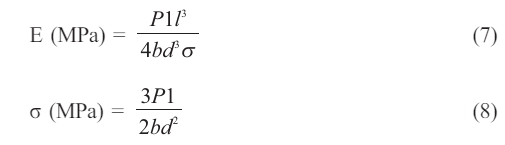
“P stands for the maximum load, l is the space between the supports, P1 is the load at the chosen elastic area point on the stress-strain plot, d is the thickness of the specimen, and b is its width”.
Compression
The compression set is assessed according to ISO 1653 at ambient temperature.
|
Table 1 Design of the study groups showing the altered SiO2 Nanofillers and altered silane (MPTMS) concentration with various monomers. |
Confirmation of silane treatment by FTIR
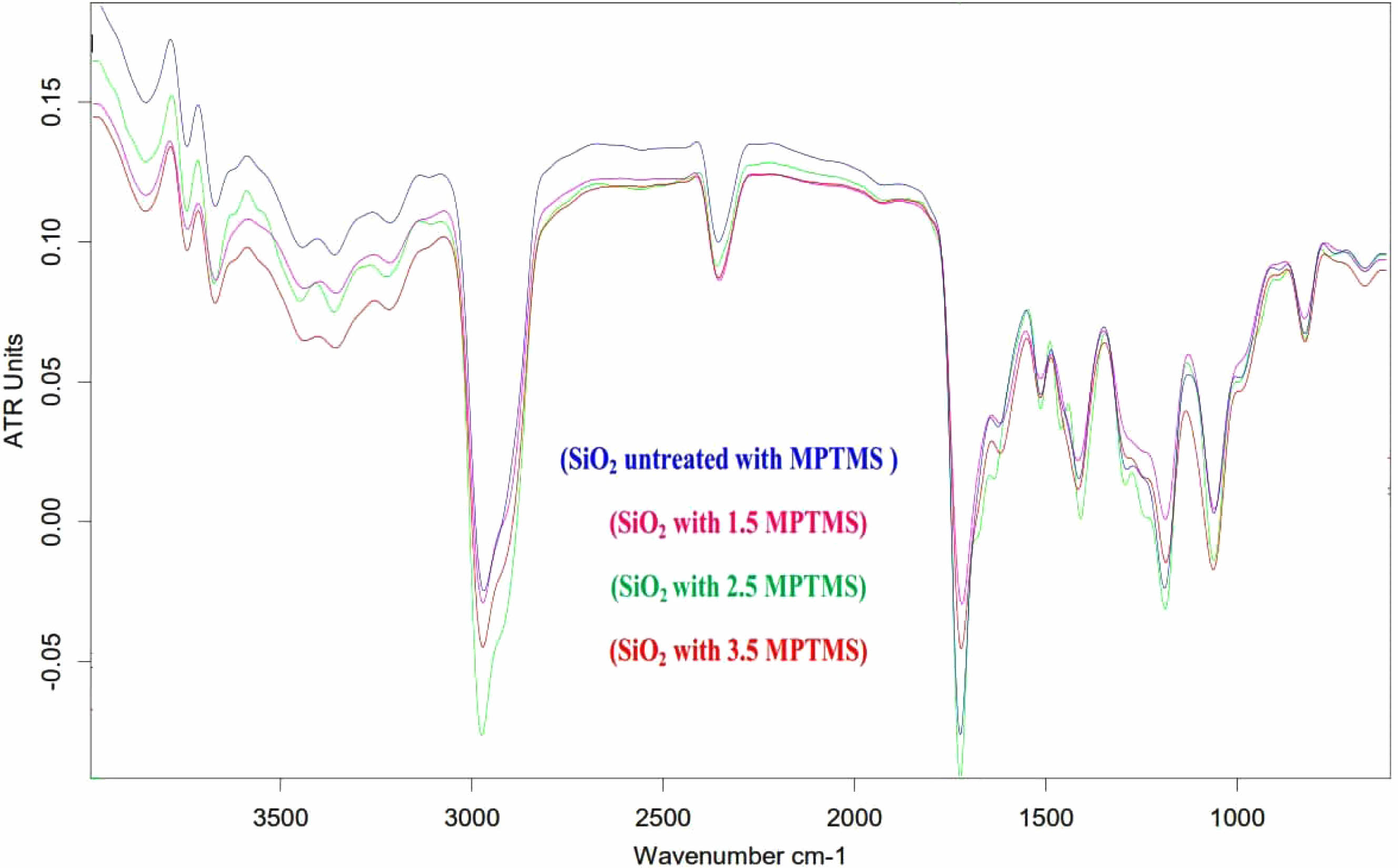
After MPTMS (1.5, 2.5, and 3.5 wt.%) “silanization of the nano-SiO2 surface, the functionalized powder is tested using Fourier transform infrared spectroscopy”. Fig. 1 shows the FTIR spectra of both the reference and the functionalized Nano-SiO2 particles. Absorption peaks at 1110 and 1654 cm-1 in Nano-SiO2 are assigned to the Si–O–Si stretching and C=O bending vibrations, respectively. The untreated particles display a doublet absorbing on 1200 and 1020 cm-1, signifying the presence of the Si–O–Si [12]. Absorption peaks near 460 cm-1 were attributed to Si-O vibrations. Unique absorption peaks at 3129 cm-1 are attributed to coupling agent-related -CH2 groups on a Nano-SiO2 surface. The MPTMS functionalized Nano-SiO2 particles provided a feeble, noticeable absorption top at 2523 cm-1, credited to the S.H. stretch, that is not noticed in the spectrum of absorption of Nano-SiO2 particles. An additional Si–O–Si peak at 1183 cm-1 appears due to the functionalization of the Nano-SiO2 surface through covalent bonds.
Synthesis and Characterization of Dental Nano Composite
Synthesis as well as FTIR spectrum of (A)
Copolymerization of Methylmethacrylate, UDMA, and Bis-GMA in the presence of 1,6-hexandimethacrylate as a crosslinking agent, C.Q. as an initiator, and (DMAEMA) as an accelerator is used to prepare dental nanocomposites (series A) in the presence of nanofillers. The reaction (Scheme 1) was done for 2 h at a temperature of 26 °C in the presence of N2 gas to remove oxygen. 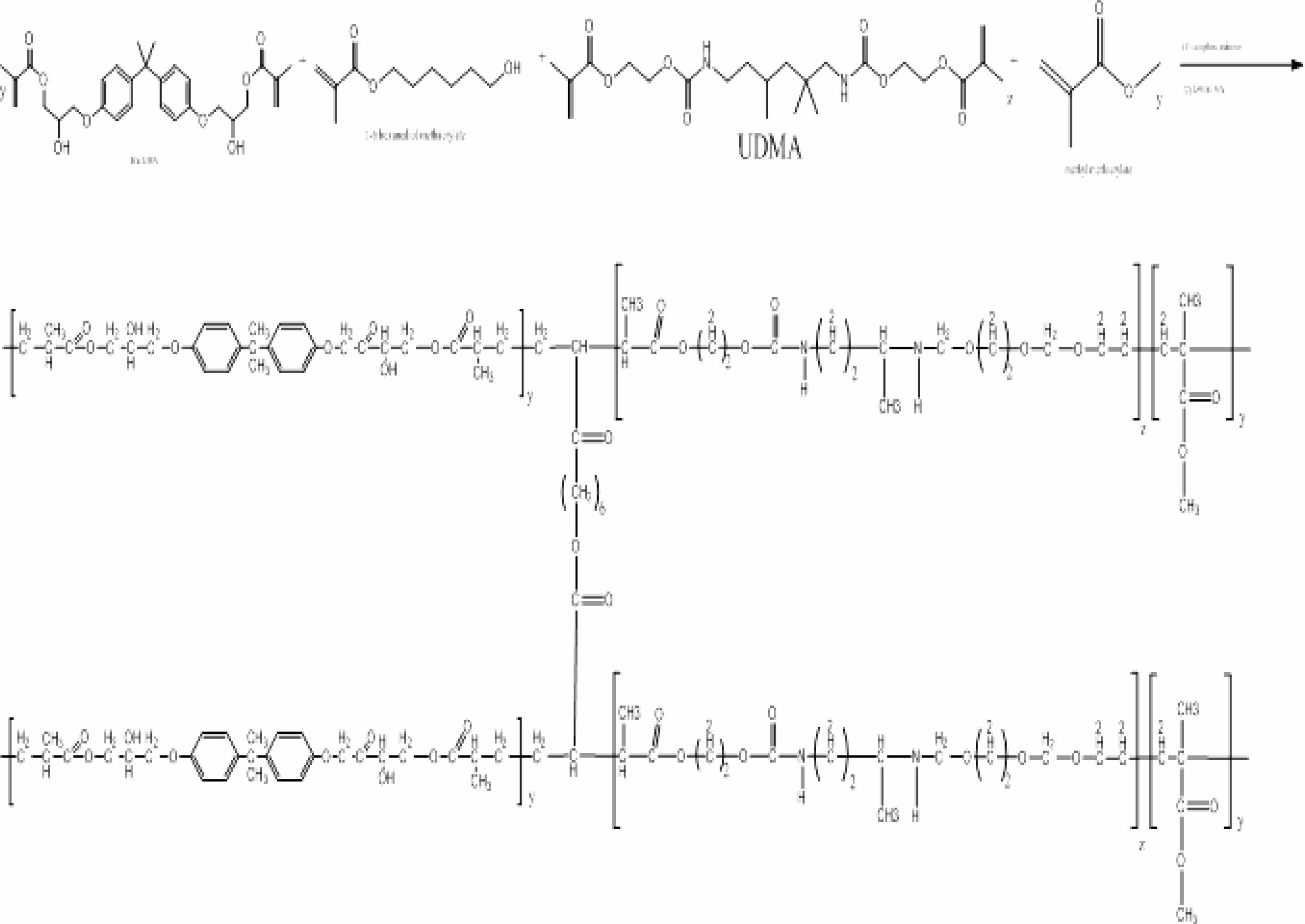
Scheme 1. Synthesis of resin during the formation of DNC (A).
FTIR spectrum analysis
The I.R. spectra showed many peaks, notably between 3200 and 3400 cm-1, which superimposes the known absorbance (O.H.). The presence of the peaks from the 2890-2980 cm-1 range is associated with vibrations of the aliphatic C-H bonds specific to the polymer. Ester group (C=O) vibration peaks may be seen at 1725 cm-1, while aromatic (C=C) peaks can be seen at 1548 cm-1.
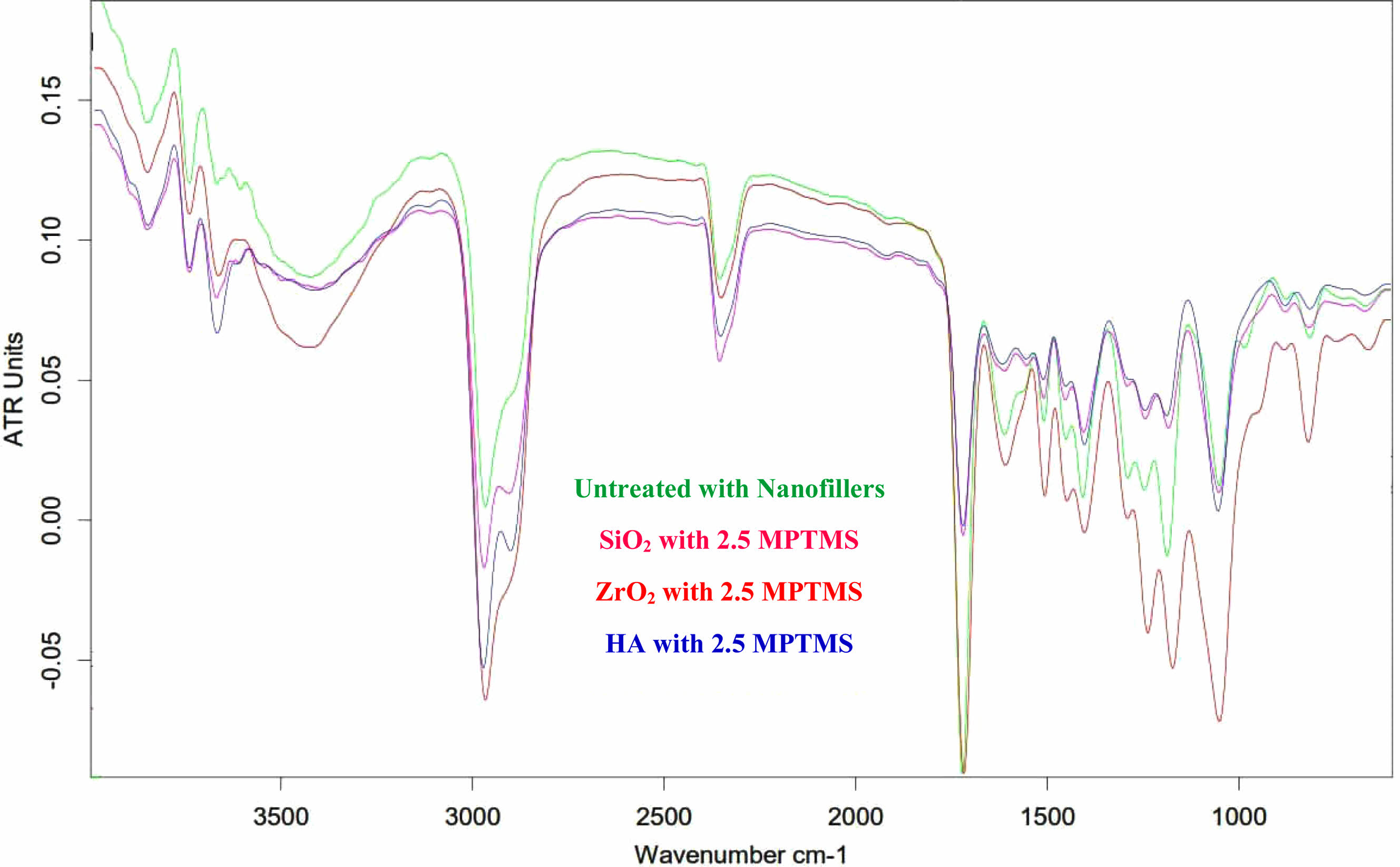
Distinctive bands in the range of (1375-1450 cm-1) are the outcome of (C–H) bonds to the (CH3) group of polymers. C-O vibrations were present, as indicated by peaks between 1000 and 1100 cm-1, which were superimposed on silica vibrations. PO4-3 group peaks at 560 as well as 600 cm-1 as well as at 1000-1100 cm-1. CO3-2 group shapes feeble peaks amongst 870 as well as 880 cm-1 as well as additional intensive peaks amongst 1460 as well as 1530 cm-1. The peaks on 2360 cm-1 designate the hydroxyl zirconium (Zr–O.H.) bond stretching vibration [13]. Sharp peaks at about 500 cm-1 conform to the Zr–O of stretching vibration ZrO2. The MPTMS functionalized Nano-SiO2 particles provided a soft, noticeable absorption peak at 2523 cm-1, credited to the S.H. stretch, that is not noticed in the spectrum absorbance of Nano-SiO2 particles. A Si–O–Si peak transmittance on 1183 cm-1, as well as C=O vibration greatest at 2430 cm-1, were established to deal with SiO2, denoting the agent that is coupling being bonded on the Nano-SiO2 surface via covalent bonds. This displays that the immobilization of MPTMS onto Nano-SiO2 was fruitful. In the condition of the functionalization of silane, FTIR spectra showed a robust peak of absorption on 1725 cm-1 as well as two other peaks on 2935 as well as 3477 cm-1. The peak absorption on 1725 cm-1 agrees with the group of carbonyls extending way in MPTMS [14, 15, 16]. as may be realized in Fig. 2. Fig. 3
Synthesis and Characterization of Dental Nano Composite
Synthesis and FTIR spectrum of DNC(B)
The process of preparing Dental Nanocomposite B (denoted DNC B) is done through a copolymerization process which is connected with the Methacrylic acid, bisphenol A dimethacrylate, Bis-GMA, with a cross-linker agent (1,6-Hexandimethacrylate), (CQ) as initiator as well as (DMAEMA) as an accelerator. The reaction was continuous over a procedure of mixing for two hours at the temperature of 26 °C below N2 gas to eliminate the oxygen that is dissolved, only as may be witnessed in reaction Scheme 2.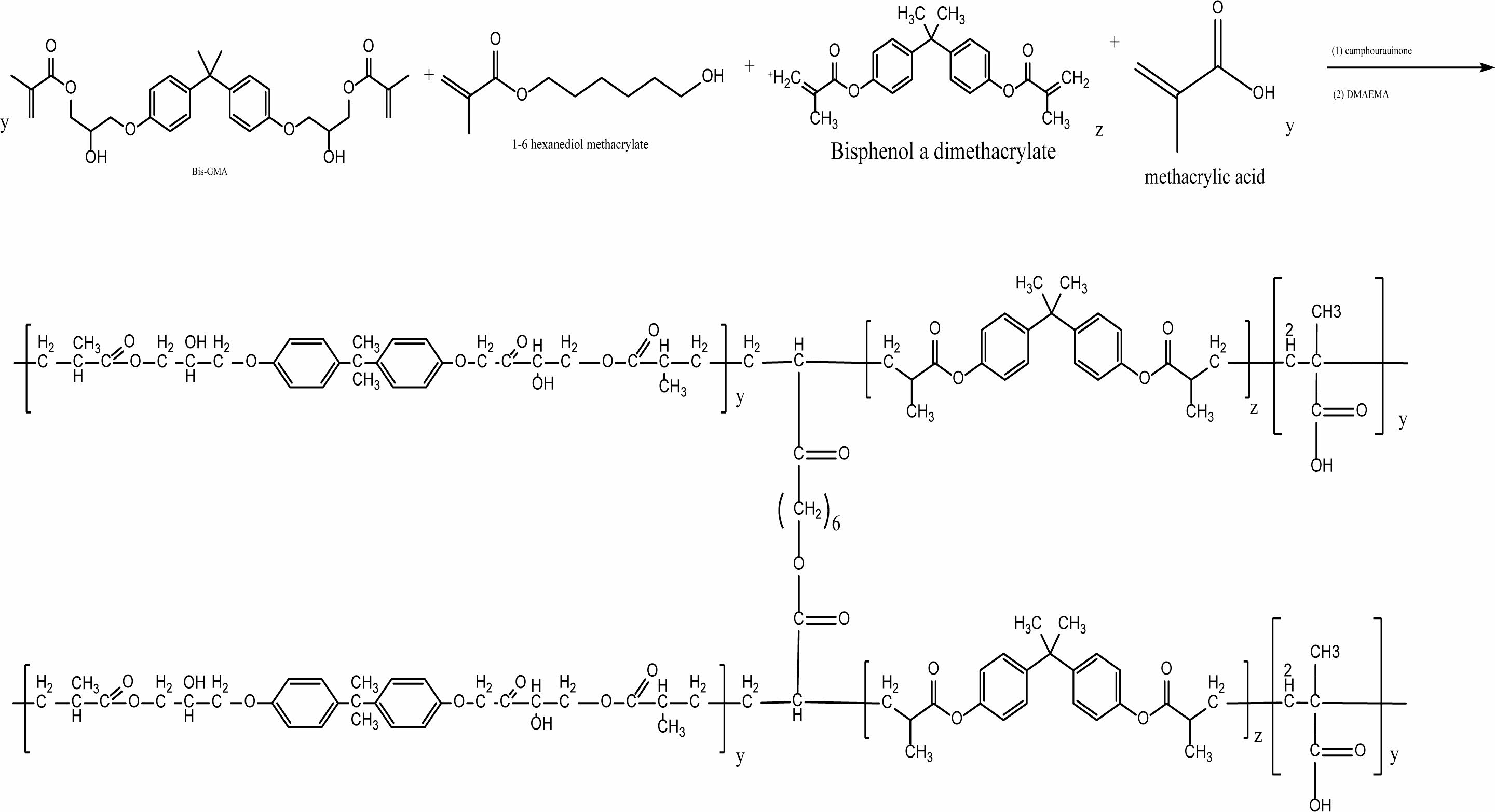
Scheme 2. A synthesis scheme of the resin of DNC (B).
FTIR spectrum analysis
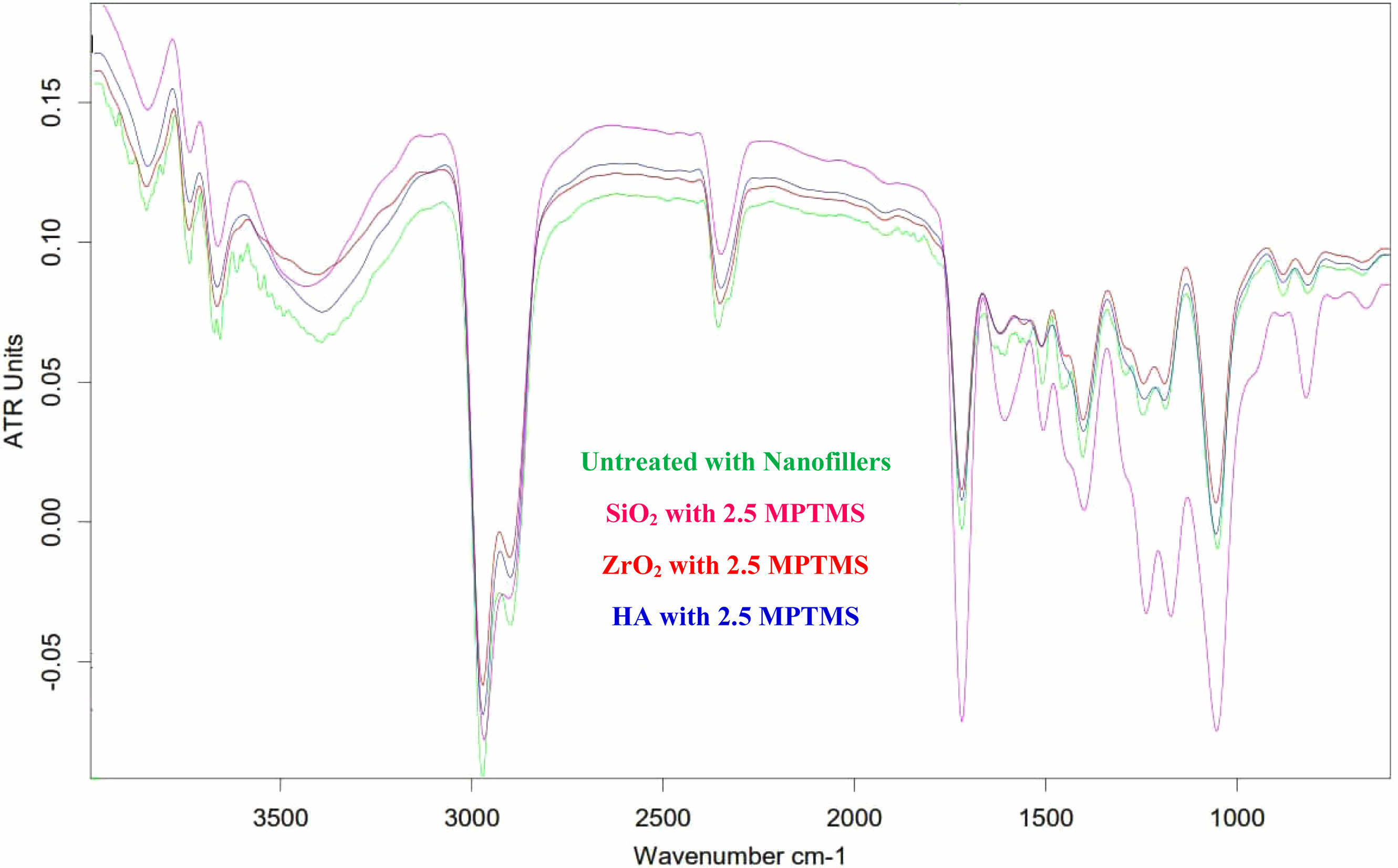
The infrared spectrum displays several peaks, the most prominent of which is the broad peak at (3200-3400) cm-1, which superimposes itself on the already-present absorption feature (O.H.). The peaks that appeared ranging in (2890-2980 cm-1) are connected to aliphatic bonds of C.H. in the structure of the polymer—distinguishing peaks by 1725 cm-1 designate (C=O) vibrations regarding the ester group. Additionally, aromatic (C=C) vibrations were designated at (1548 cm-1). Characteristic bands in the rate of (1375-1450 cm-1) were the outcome of (C–H) bonds related to the (CH3) group of polymers. The peak’s appearance ranging in (1000-1100 cm-1) indicates (C–O) vibration. PO4-3 group peaks at 560 as well as 600 cm-1 as well as at 1000-1100 cm-1. CO3-2 group shapes feeble peaks amongst 870 and 880 cm-1 and many stretch peaks amongst 1460 and 1530 cm-1. The peaks at 2360 cm−1 designate the vibration that is stretching of the hydroxyl zirconium (Zr–O.H.) bond [13]. Sharp peaks at about 500 cm-1 conform to the vibration stretching of Zr–O in ZrO2. The MPTMS functionalized Nano-SiO2 particles provided a weak, however noticeable absorbance highest at 2523 cm-1, credited to the Si–H stretch, that is not noticed in the spectrum of absorption of Nano-SiO2 particles. A Si–O–Si peak transmittance by 1183 cm-1, as well as a C=O vibration peak of 2430 cm-1, are established to deal with SiO2, denoting the agent that is coupling being bonded on the Nano-SiO2 surface through covalent bonds. This displays that the immobilization of MPTMS onto Nano-SiO2 was fruitful. Regarding the functionalization of silane, FTIR spectra showed a robust absorption top at 1725 cm-1 and two other peaks at 2935 and 3477 cm-1. The absorbance peak at 1725 cm-1 agrees with the carbonyl group extending way in MPTMS [14, 15, 16]) as seen in Fig. 4.
Scanning electron microscopy (SEM)
The SEM fractured surfaces’ images corresponding to the dental nanocomposites obtained with treated and untreated fillers are given in Fig. 4. The surfaces of fractured DNC are rough and homogeneous regardless of the used functionalized nanofillers (SiO2, ZrO2, and H.A.). This means that the functionalization is beneficial and helps their dispersion into the polymer matrix.
Figs. 4-5 highlight that the functionalized nanofillers are well dispersed into the polymer matrix, and the size of the nanoparticles is around 40 nm. Also, these images prove a better adhesion between the organic and inorganic phases. Also, significant agglomerations are visible while the distance amongst nanofiller particles (SiO2, ZrO2, as well as H.A.) is significantly larger than the size of the nanoparticles, thus proving good compatibility between the phases again, and the mechanism of fracture involves instead a polymer fracture instead of the polymer detaching from the surface of the nanoparticles. There are areas with high filler agglomerates. However, areas that are mainly polymer and these heterogeneities can substantially impact the mechanical properties. Overall, based on the SEM evaluation, surface modification with MPTMS is beneficial and leads to better dispersibility and compatibility within the polymer.
Better mechanical properties in dental composites can be achieved by establishing a solid covalent link between the inorganic fillers and the organic polymer matrix, as demonstrated by Chen (2010) [17]. As a coupling agent, silane possesses the right kinds of groups to join the filler to the matrix chemically. Therefore it can be used to join the two phases together. Because of the Acrylic nature of the polymer matrix, in this paper (A characteristic agent that is coupling MPTMS was chosen as a silanization/coupling agent like manipulated in this work). For instance, the MPTMS molecule can be attached to the inorganic fillers via their hydroxyl groups of the silanolic groups. At the same time, the acrylic moiety is available for copolymerization with the polymer matrix of the acrylic monomers. This way, a true covalent bond is realized between the resin and the filler.
Fig. 4 displays the SEM images of the dental composites obtained with the three salinized nanofillers and the polymer composition belonging to the DNC A series. These nanofillers are assembled in a bunch-like structure, embedding the spherical nanofillers (SiO2, ZrO2, HA) of 32-38 nm. SEM micrograph of each material displayed that constructing a robust bond covalent amongst inorganic fillers and the organic matrix is vital for gaining mechanical properties in dental composites. Connecting these phases is attained by treating the fillers with the coupling agent – MPTMS, which, owning adequate functional groups, can connect the filler and the matrix via a chemical bond. Both the hydroxyl groups of the silica particles and the polymer matrix can benefit from one outcome of the molecule’s reaction.
Fig. 5 illustrates the distinct morphologies of composite materials based on DNC B and three nano-functionalized fillers (SiO2, ZrO2, HA). Similar to the SEM pictures acquired for the DNC A series, these SEM images reveal the spherical structure of the nanoparticles embedded inside the polymer matrix. In addition, there is no visible aggregation of these nanoparticles, indicating that their dispersion was successful and that silanization of the nanofillers is likely significant, as we may deduce when comparing these pictures to those obtained with composite samples containing unmodified nanofillers.
Water Sorption (W.S.)
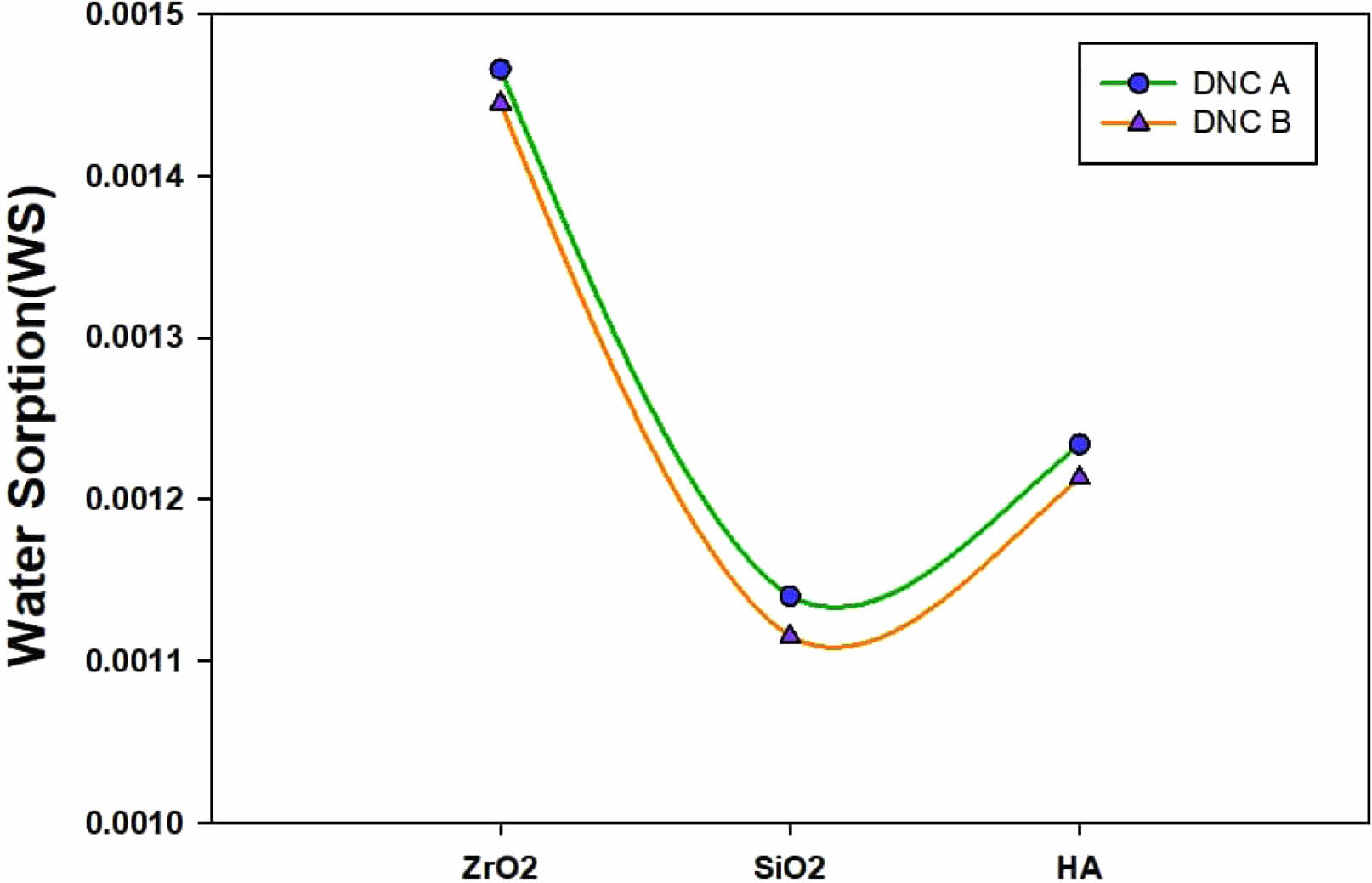
The chemical composition of the prepared composites materials, including the ratio between the hydrophobic and hydrophilic monomers, solvent, nanofiller but also functionalizing agent (MPTMS), has a straight influence on the overall performance of the restorative dental nanocomposites. Superficially, the degree and the rate of the water’s absorption are related to the strong hydrogen bonding and dipole interactions, as proven in [18].
“Because of the swelling when water diffuses through polymeric materials, it reduces the frictional forces between the polymer chains, which is undesired. The polymeric chains may relax upon increased water absorption, allowing the escape of unreacted monomers and/or solvents trapped inside the polymer network [19]. These factors are probably the reason for the recognized degradation of bonds that are resin-dentin over time noticed by several researchers” [20, 21].
Water sorption has a significant effect on the durability of dental material appliances in aqueous environments, such as the mouth, because water intrusions can cause a variety of unfavorable effects, such as the hydrolysis of polymeric networks [22], a decrease in thermal stability [23], a degradation of the mechanical properties [24], and the release of unreacted monomer [25].
Regarding the present study, there is a rise in water sorption using the reduction in conversing the tested formulation. Due to the character of the molecule’s hydrophobic, and the raised conversion, Bis-GMA is given thru the lowest sorption of water [24]. In concern to the 1,6-hexandioldimethacrylate, conversions that are elevated do not interpret to the raised density of crosslinking, being mentioned previously, therefore second to last lowermost water absorption is justly unexpected [26], it should too be related to its small hydrophilicity compared to Bis-GMA. Very comparable values of water sorption are exposed through hydrophobic monomers as well as Bis-GMA, despite dissimilarities in the conversion, perhaps because of the truth that these are regarded to be the main hydrophilic molecules measured in this work.
It should also be demonstrated that the results of solubility may be undervalued for more hydrophilic monomers due to the water’s yielding hydrogen bond that is strongly interacting with the hydroxyl and (in Bis-GMA), a weak bond using the ethylene glycol unit (which was offered in higher concentrations in 1,6-hexandioldimethacrylate), which may be preventing the water removal during the storage period in desiccators [27].
It may be demonstrated that Bis-GMA-dependent resin possessed the same properties before and after water immersion, indicating its superior water resistance. Tissue inflammation and cytotoxicity can be triggered by releasing unreacted monomers from polymeric networks, which is revealed by solubility [28]. Because oxygen atoms have higher electronegativity, and cohesive energy, hydrogen bonds formed between molecules via -N.H. are weaker than those formed by -OH [29]. Bis-GMA-based polymers may have poor solubility because unreacted monomers are more likely to be absorbed by surrounding networks and then released from the polymers. Bis-GMA monomers form many hydrophobic molecules; therefore, the materials shall show minor sensitivity to the liquid’s sorption, as demonstrated in Fig. 6.
As the polymethacrylates may be regarded as crosslinked dental polymers, as well as due to the chains of a polymer having crosslinks amongst them, such will characteristically cause a significant reduction in solvent permeability of the polymer since the decrease in the ability to connect to a chain of the polymer for swelling and dropping hole free volume [30]. However, water sorption may be clearly described as a desired result as the expansion of the composites’ hygroscopic is designed to cover the marginal leakage gap caused by the shrinkage [31]. Meanwhile, a special requirement for dental composites is adequate resistance to deterioration by water and other solvents.
It is expected that, after 30 days, just 0.02% to 0.05% of the total mass of dental composites will have leached into the aqueous solution as a result of inorganic ion migration (Nano-fillers).
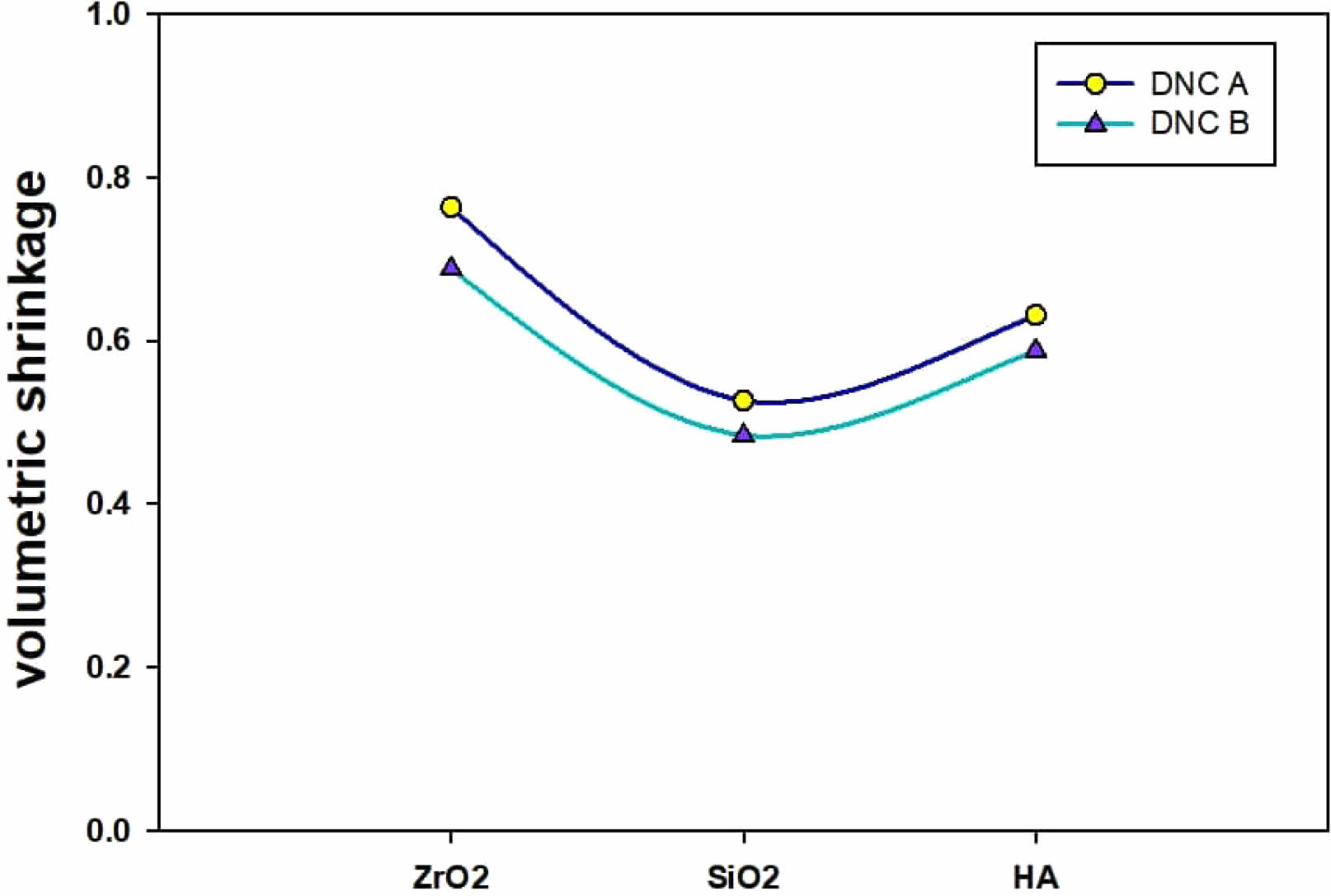
Volumetric Shrinkage
According to the Archimedes principle, we may calculate the Volumetric Shrinkage (VS) values associated with the composites’ specimens. After allowing an uncured sample to rest for one minute, the slumping impact on measurements can be eliminated by subjecting the sample to 40 seconds of light curing with the curing unit’s tip positioned at a distance of (1-2 mm) from the specimen. The Commercial Density Determination Kit of a Mettler Toledo X analytical balance is used for the evaluation by ISO17304:2013 standards (E).
The composite material’s polymerization has been followed by volume reduction that can be considered a contraction stress at the tooth’s contact. One of the critical drawbacks, mainly in dental composites, is that their polymerization shrinkage might result from secondary caries and marginal gaps in restored teeth. After the polymerization process begins, and as it continues, van der Waals distances are converted into covalent bond lengths, resulting in a reduction in volume. “The degree of contraction can be represented by the number of newly formed covalent bonds, also known as the degree of the polymerization reaction. Another reason for volume reduction is that the molecular distances between polymer chains are smaller than between monomers” [32]. As a result of the pressures caused by polymerization shrinkage, tooth-restorative interface debonding can occur in relatively short amounts of time [33]. A significant challenge in developing dental composites is to remove or reduce polymerization shrinkage.
Volumetric Shrinkage’s magnitude recorded for the composites is determined by the action of filler volume and the degree of conversion as well as the composition of the resin, which is revealed in Fig. 7.
A different study concluded that great ratios of 1,6-hexandioldimethacrylate/Bis-GMA within the composite materials would show great contraction stress value due to the higher VS because of the developed conversions [30]. As a result of the “diluent” monomers increasing the density required to polymerize carbon double bonds, the host monomers will shrink more rapidly than they otherwise would. Because of the elevated Tg and decreased viscosity of the diluent, the rate of the reaction environment would increase, allowing for more powerful conversions [34]. Before the curing process, monomers with a large chain like Bis-GMA and 1, 6-hexandioldimethacrylate have been loosely dispersed in the matrix. Afterwards curing, the reaction with monomers happened. Amounts of crosslinked points did not accompany the chains ‘ movement, and the free volume of resins was altered remarkably. This outcome resulted in an outstanding decrease in volumetric shrinkage. Nanofillers have a structure of a three-dimensional organic-inorganic hybrid cage structure, and their volume is much greater than those of Bis-GMA and 1,6-hexandioldimethacrylate. When nanofillers are incorporated into the matrix, they may attract and even entwine the monomer chains, and the monomers may disperse more firmly. Nanofillers restrict the change of the matrix’s free volume throughout the reaction, and the nanocage of nanofillers does not alter the reaction afterwards. Briefly, the shrinkage of the volume of dental composites loaded with nanofillers decreases.
Mechanical properties
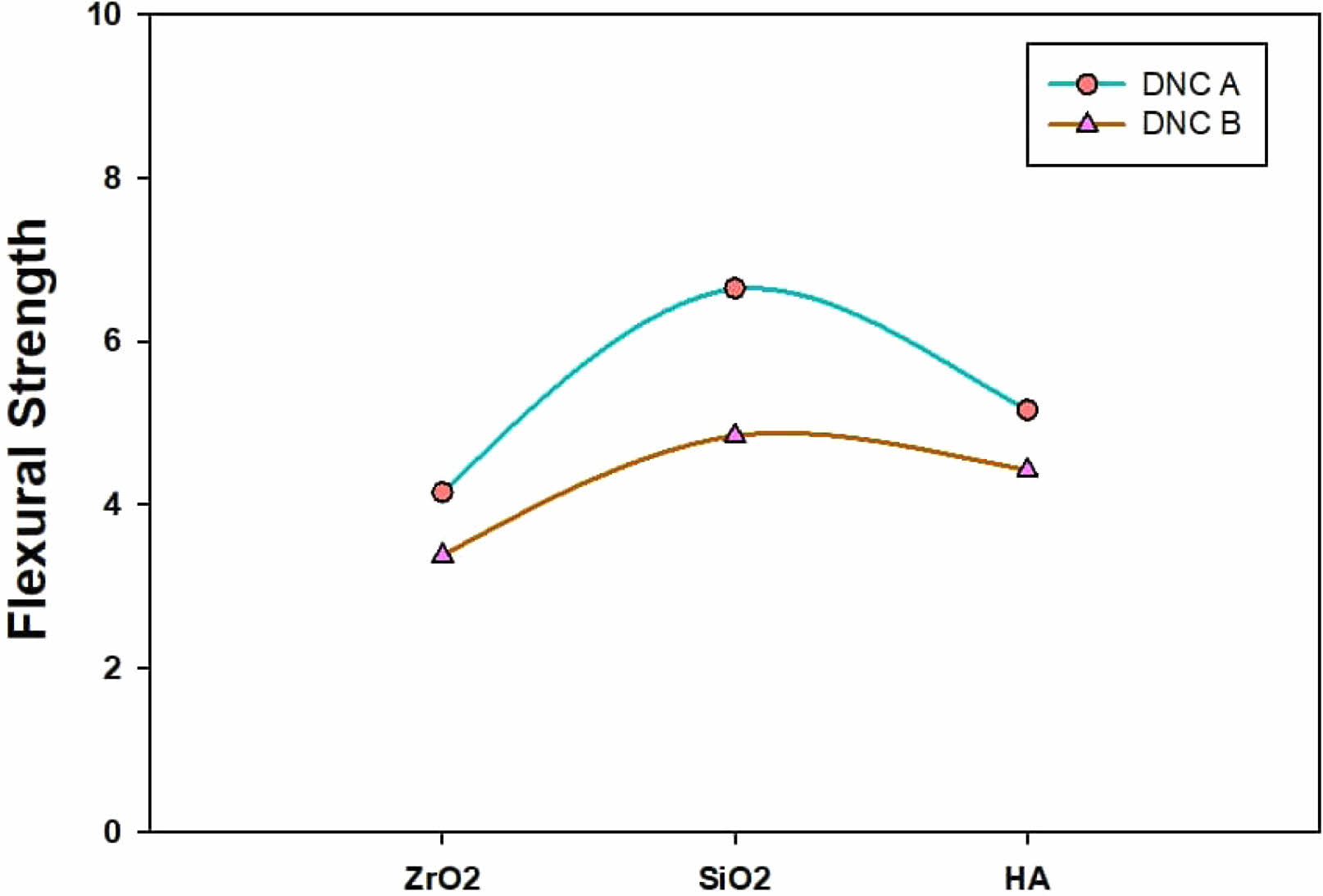
Because it includes the effects of both compressive deformation (around the load’s application point) “and tensile deformation, the flexural test is widely employed in dental research to examine the mechanical behaviour of dental composites” (on the contrasting specimen side). Both dental nanocomposites displayed satisfactory flexural strength values that comply with ISO standards.
The resultant composites’ mechanical characteristics typically increase when more filler is incorporated. In several recent investigations, mechanical parameters (flexural strength, compressive strength, hardness, and elastic modulus) have been shown to increase with increasing filler amounts [35, 36, 37]. Dental composites, especially those labeled nanocomposite, typically include far larger loads of fillers of the micron size than the nanofillers used in research. Therefore, roughly 70-80 wt.% of filler can be used to fill them. However, the fillers used in this study are all nanometric in size (SiO2, ZrO2, HA), and they demonstrated a high surface area even at filler loadings of 10 wt.%.
Group DNC A gave a faintly greater mean flexural strength than group DNC B. The two composites had the same kind of monomer resin as well as fillers, as well as the only dissimilarity was the filler’s content. The combination of overloaded filler content perhaps dropped the flexural strength, which agrees with the outcomes of other studies [38, 39]. According to Adabo et al. [40], a highly packed material has a higher elastic modulus but is more prone to tiny fractures.
The elastic modulus, which evaluates the composite’s capacity to flex under stress, is another critical mechanical characteristic established by the flexural test [41]. It is possible that mechanical stress could cause a composite to break after it has become very rigid. However, if it is exceptionally malleable, it may not have survived the stresses experienced in the mouth. The composite’s elastic modulus increases as more fillers are incorporated. Depending on the outcomes attained, the modulus of group DNC A was significantly greater than that of group DNC B.
Despite being the composite’s weakest link, the filler/matrix interface is crucial in determining the material’s mechanical properties. Because of the solid filler-matrix connection induced by the coupling agent, fracture propagation is slowed down in the polymer matrix. [42]. “In contrast, with the unmodified composites, the fracture is frequently initiated and propagated via flaws at weak polymer-filler particle interfaces. Generally, the extensibility of the samples is greater compared with the systems with non-modified fillers, though it can be affected through the testing samples’ surface conditions” [43]. Mechanical properties for all the compositions of dental nanocomposite are presented in Figs. 8-9. It may notice that the flexural strength raised progressively to 10 wt.% of the filler loading in the presence of the coupling agent. These outcomes are connected to the filler’s interaction with the matrix. When functionalized nanofillers are used, the derived nanocomposites displayed positive outcomes by developing strength [44]. The highest values in these composites are obtained at 10 wt.% Nanofiller. In general, silanization/coupling agents can help one achieve optimal mechanical properties while using a smaller volume of filler (especially if the filler is in the nanometric range). The coupling agent facilitates improved performance through its role as a bridge between the filler and the matrix.
Based on the flexural strength (Fig. 8), it is confirmed that the efficiency of the treatment presents an optimum. For instance, 2.5% concentration yields maximum flexural strength for 10 wt.% nanofiller for both polymer compositions. For 10 wt.% contents of the filler, the trend is too apparent where the flexural strength rises by rising concentration from 1.5 to 3.5% is the highest at 2.5 wt.%.
We cannot draw any firm conclusions about the relationship between flexural strength and treatment concentration for the 15 wt.% filler system. In general, the development of specific properties results from the drop in the filler’s hydrophilicity as an outcome of their treatment, making the fillers much more well-matched by the hydrophobic [45-47].
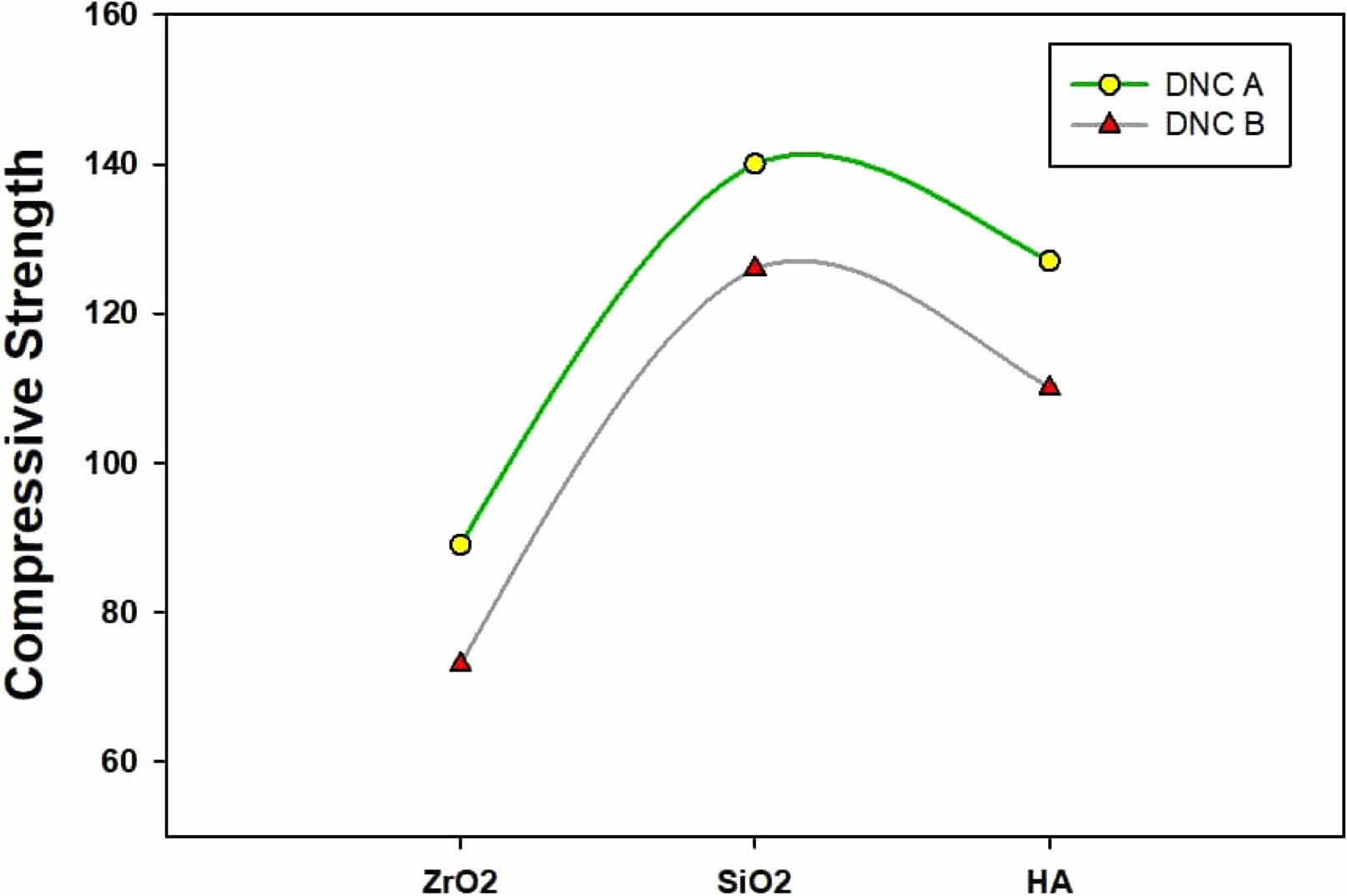
The compressive strength of the two series of dental composites is shown in Fig. 11. Results show that the compressive strength of Group A is higher than that of Group B, the highest values being for the samples loaded with functionalized SiO2 (150 MPa for the DNC A compared with ~130 for the DNC B).
Besides the nature and the filler’s size, the resins play a substantial role in developing improved strength. The presence of aromatic cycles of the Bisphenol di-methacrylate and Bis-GMA is noticed in dental composites outcomes in low cyclization capacity and rising crosslinking in the polymer as deliver consecutive improvement in the mechanical features such as strength and durability. However, due to its high adaptability, intermolecular cyclization is common in acrylate and UDMA monomers. Bis-GMA rigidity plays an essential role in the attainment of high compressive strength [48, 49].
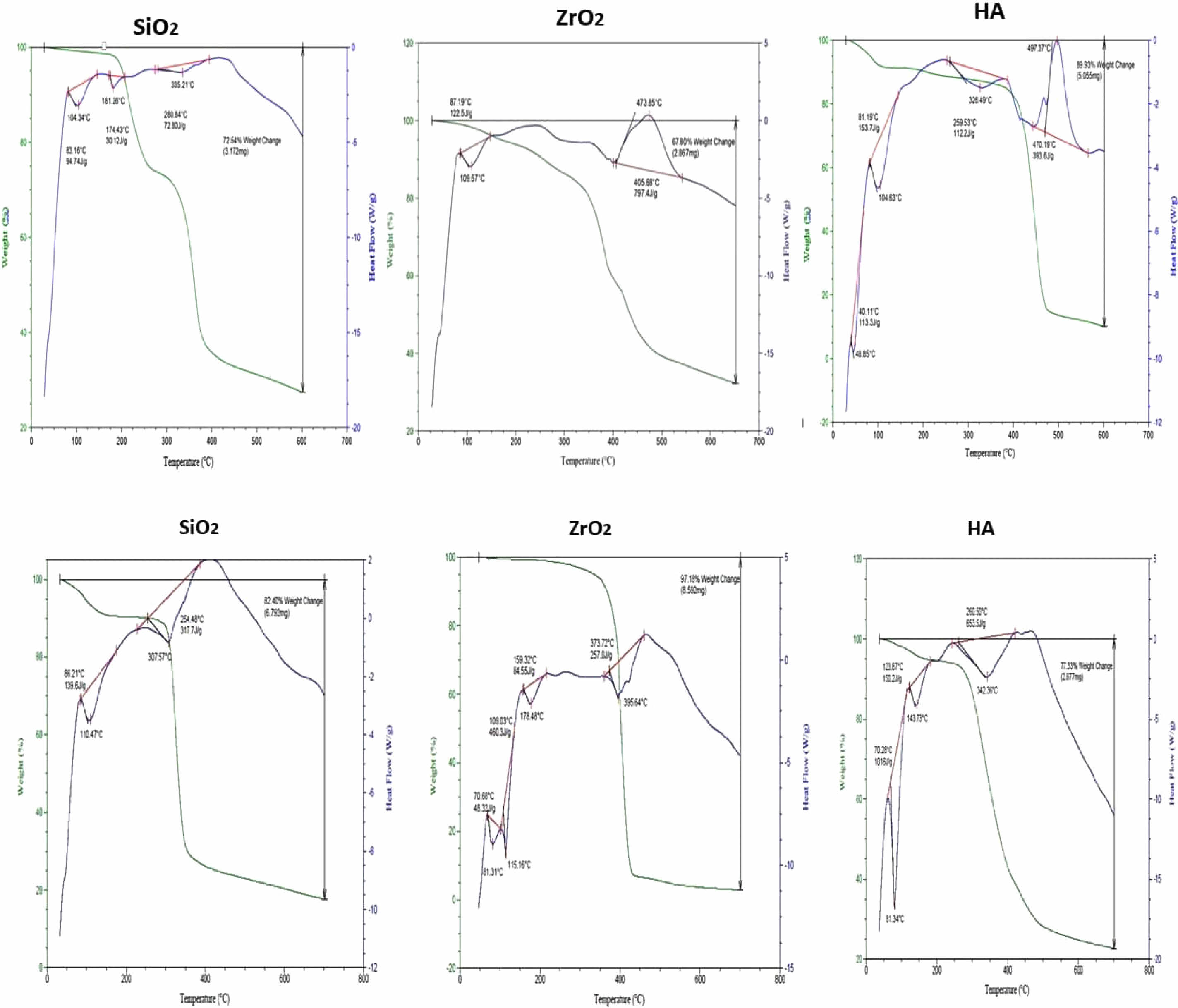
Thermal Gravimetric Analysis (TGA) Study
“Ti, Top, Tf, T50%, % Residue at 600 ºC, as well as char yields at 500 ºC are presented in” (Table 4).
Both DNC A and DNC B had 50% weight loss temperatures between 360 and 490 degrees Celsius; the char results for DNC A were 15 to 33%, and those for DNC B ranged from 7 to 28% at 500 degrees Celsius in an Argon atmosphere, indicating that they could be used as temperature-resistant supplies for a variety of purposes. At 600 oC, DNC A has a weight residue of 10-27%, while DNC B has a 5-25% residue.
“Differential Scanning Calorimeter Analysis DSC Study”
Based on Fig. 10, the transition of glass value (Tg) of the DNC a group is between 104.34 and 128.95 °C) representing that the flow of temperature as well as after that rise in the absorption rate of the sample to every point of melting (Tm) is between (440-473 °C) is dissolved as well as after that the absorption rate of the sample to the heat as well as thru the curve is set the crystallization temperature (Tc) of the nanocomposite (335.21-497.37 °C).
Based on Fig. 10, the essential thermo-gravimetric characteristics of the DNC B were determined, including the glass transition temperature (Tg) of the dental nanocomposite is 81.31 and 110.47 °C; the melting temperature (Tm), which is between 440 and 480 oC and the temperature of crystallization (Tc) of the nanocomposite between 307.57 and 395.64 °C. Table 5
|
Fig. 1 FTIR spectra of the untreated as well as treated Nano-SiO2. |
|
Fig. 2 FTIR spectra of the treated Nano-fillers for DNC (A). |
|
Fig. 3 FTIR spectra of the treated Nano-fillers for DNC (B) |

|
Fig. 4 SEM image of the dental composites belonging to Group A and modified nanofillers (SiO2, ZrO2, HA). |

|
Fig. 5 SEM image of the dental composites belonging to Group B loaded with functionalized nanofillers (SiO2, ZrO2, HA). |
|
Fig. 6 Water sorption (WS). |
|
Fig. 7 Volumetric Shrinkage. |
|
Fig. 8 The dental nanocomposite materials’ flexural strength (MPa) loaded with 2.5 wt.% MPTMS-based modified. |
|
Fig. 9 The compressive strength (K.N.) of the dental nanocomposite materials loaded with 2.5 wt.% MPTMS-based modified nanofillers. |
|
Fig. 10 Curves of TGA and DSC for the DNC A and DNC B series. |
|
Table 4 Some Thermo-Gravimetric Characteristics of the DNC (A and B). |
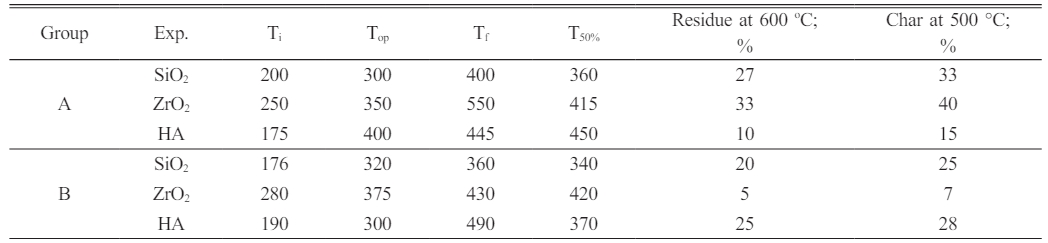
D.T.: “Decomposition temperature”. |
|
Table 5 Glass Transition Temperature, Melting Temperature as well as the Temperature of Crystallization as determined by Differential Thermal Analysis of the dental nanocomposites. |
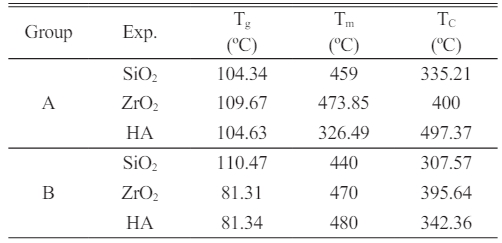
Tg: Degree of the transition of glass |
The use of nanoparticles (SiO2, ZrO2, HA) as reinforcing agents for dental applications has been intensively explored. There are several commercially available materials whose performance might be significantly enhanced by modifying their size and surface. The SEM micrographs and reflectance data indicate that functionalizing the nanofillers (SiO2, ZrO2, HA) might result in better interaction between the polymer matrix and the nanometric filler, but also in their aggregation. Due to the improved compatibility between phases, the functionalized nanofillers dramatically modify the crystallinity of the composite material. In this study, 3-(methacryloyloxy)propyltrimethoxysilane (MPTMS) was utilized as a silanization agent due to the methacrylic moiety’s ability to form chemical bonds with methacrylic monomers. The mechanical characteristics of these nanocomposites for dental applications were evaluated and found to be appropriate for dental applications. The flexural strength of composites is highly dependent on the degree of adhesion between filler and matrix. The surface of the filler was changed using MPTMS, resulting in excellent mechanical properties and exceptional wear resistance. Despite the limitations of the investigation, it is feasible to make conclusions regarding the advantages of the synthesized dental nanocomposites, such as increased water resistance and decreased volumetric shrinkage.
- 1. K. Anusavice, Adv. Dent. Res. 17[1] (2003) 43-48.
-

- 2. K. Leinfelder, J. Am. Dent. Assoc. 128[5] (1997) 573-581.
-
- 3. E. Yoonis and M. Kukletová, Scr. Med. 82[2] (2009) 108-114.
- 4. Swift Jr., E. J., Esthet. Restor. Dent. 22[1] (2010) 72-77.
-
- 5. S. Mitra, D. Wu, and B. Holmes, J. Am. Dent. Assoc. 134[10] (2003) 1382-1390.
-
- 6. B. Kahler, A. Kotousov, and K. Borkowski, Dent. Mater. 22[1] (2006) 942-947.
-
- 7. N.J. Opdam, J.J. Loomans, F.J.J. Roeters, and E.M. Bronkhorst, J. Dent. 32[5] (2004) 379-383.
-
- 8. J. Ferracane, Dent. Mater. 22[8] (2006) 689-692.
-
- 9. D. Behera and A. K. Banthia, Adv. Mater. Res. 29-30 (2007) 241-244.
-
- 10. Y. Alexander Fadeev, H. Roy, and M. Stevan, Langmuir. 18[20] (2002) 7521-7529.
-
- 11. M. Chen, J. Dent. Rest. 89[6] (2010) 549-560.
-
- 12. D. Jones and A. Rizkalla, Mater. Res. Appl. Biomater. 33[1] (1996) 89-100.
-
- 13. K. Koshiro, S. Inoea, T. Tanaka, K. Koase, M. Fujita, M. Hashimoto, and H. Sano, Eur. J. Oral Sci. 112[4] (2004) 368-375.
-
- 14. J. Ferracane, Dent. Mater. 22[3] (2006) 211-222.
-
- 15. R. Osorio, J. Pisani-Proença, M.C. Erhardt, E. Osorio, F.S. Aguilera, F.R. Tay, and M. Toledano, J. Dent. 36[2] (2008) 163-169.
-
- 16. K. Soderholm, J. Dent. Mater. 26[2] (2010) 63-77.
-
- 17. A.M. Costella, J.L. Trochmann, and W.S. Oliveira, J. Mater. Sci. Mater. Med. 21[1] (2010) 67-72.
-
- 18. I. Sideridou, V. Tserki, and G. Papanastasiou, Biomaterials. 24[4] (2003) 655-665.
-
- 19. M. Lemon, M. Jones, and J. Stansbury, J. Biomed. Mater. Res. Part A. 83[3] (2007) 734-746.
-
- 20. C. Pfeifer, Z. Shelton, R. Braga, D. Windmoller, J. Machado, and J. Stansbury, Eur. Polym. J. 47[2] (2011) 162-170.
-
- 21. J.W. Stansbury and J. Antonucci, Dent. Mater. 15[3](1999) 166-173.
-
- 22. M. Chang, L.D. Lin, F.H. Chuang, C.P. Chan, T.M. Wang, J.J. Lee, and J.H. Jeng, Acta Biomater. 8[3] (2012) 1380-1387.
-
- 23. N. Yamazaki and S. Kurata, Dent. Mater. J. 30[1] (2011) 103-108.
-
- 24. M. Giannini, M. Di Francescantonio, R. Pacheco, L.C. Boaro, and R.R. Braga, Oper. Dent. 39[3] (2014) 264-272.
-
- 25. N. Ilie, K.H. Kunzelmann, and R. Hickel, Dent. Mater. 22[7] (2006) 593-601.
-
- 26. S.H. Dickens and B.H. Cho, Dent. Mater. 21[4] (2005) 354-364.
-
- 27. C Santos, R.L Clarke, M Braden, F Guitian, K.W.M Davy, Biomaterials. 23[10] (2002) 1897-1904.
-
- 28. J.L. Ferracane and E.H. Greener, J. Biomed. Mater. Res. 20[2] (1986) 121-131.
-
- 29. B.S. Dauvillier, M.P. Aarnts, and A.J. Feilzer, Dent. Mater. 19[2] (2003) 277-285.
-
- 30. I. Ikejima, R. Nomoto, and J.F. McCabe, Dent. Mater. 19[3] (2003) 206-211.
-
- 31. S. Beun, T. Glorieux, J. Devaux, J. Vreven, and G. Leloup, Dent. Mater. 23[1] (2007) 51-59.
-
- 32. K. Masouras, N. Silikas, and D.C. Watts, Dent. Mater. 24[7] (2008) 932-939.
-
- 33. M. Tian, Y. Gao, Y. Liu, Y. Liao, N.E. Hedin, and H. Fong, Dent. Mater. 24[2] (2008) 235-243.
-
- 34. G.L. Adabo, C.A. Cruz, R.G. Fonseca, and L.G. Vaz, J. Dent. 31[5] (2003) 353-359.
-
- 35. C. Sailaja, K.T. Thilagham, K.T. Anand, P. Ganeshan, S. Kannan, A.H. Seikh and A. Ghosh, J. Ceram. Process. Res. 24[4] (2023) 617-625.
-
- 36. A van der Wal, J.J. Mulder, and R.J. Gaymans, Polym. J. 39[22] (1998) 5547-5481.
-
- 37. S. Sathiyamurthy, S. Saravanakumar, N. Ananthi, and P. Devi, J. Ceram. Process. Res. 24[4] (2023) 683-692.
-
- 38. C. Bagci, J. Ceram. Process. Res. 24[4] (2023) 700-704.
-
- 39. C. Ramesh, Mohanraj Chandran, K. Chellamuthu, and A. Sivakumar, J. Ceram. Process. Res. 24[1] (2023) 120-126.
-
- 40. K. Wang, J. Wu, L. Ye, and H. Zeng, Compos. - A: Appl. Sci. 34[12] (2003) 1199-1205.
-
 This Article
This Article
-
2024; 25(2): 178-191
Published on Apr 30, 2024
- 10.36410/jcpr.2024.25.2.178
- Received on Sep 6, 2023
- Revised on Oct 28, 2023
- Accepted on Oct 30, 2023
Services
Shared
Correspondence to
- Ammar Ali Husseina
-
Science and Engineering of Oxide Materials and Nanomaterials, Faculty of Chemical Engineering and Biotechnology, University POLITEHNICA of Bucharest, Gh. Polizu 1-7, Bucharest, Romania
Department of Science and Engineering of Oxide Materials and Nanomaterials, Faculty of Chemical Engineering and Biotechnologies, University Politehnica of Bucharest, 1-7 Gh. Polizu Street, 011061 Bucharest, Romania
Ministry of Education, The General Directorate of Educational in Najaf Al-Ashraf, Najaf, Iraq
Tel : 009627823117161 - E-mail: ammarali7778op@gmail.com
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr