





- Synthesis and luminescence properties of red lutetium based phosphors using a general precursor obtained by homogeneous precipitation
Xin Zhanga, Mingyang Liub, Jiao Hea,*, Jingbao Liana and Xue Zhanga
aSchool of Mechanical Engineering, Liaoning Petrochemical University, Fushun, 113001, P.R. China
bPolyester Factory of Liaoyang Petrochemical Branch Co, Liaoyang, 111003, P.R. ChinaThis article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
A general precursor of Lu2O2SO4 and Lu2O2S was synthesized by homogeneous precipitation methods using commercially available Lu2O3, H2SO4, Eu(NO3)3·6H2O, CO(NH2)2, NH3·H2O, Na2CO3 and S as the main raw materials, and then Lu2O2SO4:Eu3+ is calcined under air atmosphere at different temperatures, which simplified the previous experimental steps. On this basis, a series of Eu3+ doped Lu2O2S phosphors were synthesized by the solid phase method using the precursor as raw material by mixing sodium carbonate and sublimated sulfur. Lu2O2SO4:Eu3+ and Lu2O2S:Eu3+ quasi-spherical luminescent powder with particle sizes of about 1 μm were synthesized and their synthetic mechanism and photoluminescence properties were investigated. Finally, the synthesized products were characterized by X-ray powder diffraction (XRD), field emission scanning electron microscope (FE-SEM), Fourier transform infrared spectroscopy (FT-IR), differential scanning calorimeter (DSC-TG), photoluminescence spectroscopy (PL) and other analytical and testing methods. Studies have shown that the optimum calcination conditions of Lu2O2SO4:x%Eu3+ phosphors and Lu2O2S:x%Eu3+ phosphors are at 800 ℃ for 2 h in air and at 500 ℃ for 2 h in a closed sulphuration atmosphere, respectively. On this basis, the photoluminescence (PL) behavior of the two prepared fluorescent powders was analyzed. At approximately 618 nm and 628 nm, lutetium based oxysulfate and oxysulfide phosphors exhibit typical red emission, which is the main luminescent feature of Eu3+, originating from the 5D0-7F2 transition of Eu3+.The CIE color coordinates of the Lu2O2SO4:Eu3+ and Lu2O2S:Eu3+ phosphors correspond to (0.6573, 0.3423) and (0.621, 0.3769), respectively, and their colors are red-orange and orange light. The optimal doping concentration was determined to be 7.5%. The fluorescence lifetime of Lu2O2SO4:7.5%Eu3+ and Lu2O2S:7.5%Eu3+ phosphors obtained by homogeneous precipitation method is 2.4479 ms and 1.004 ms. The CCT values are 2886 K and 1863 K, which belong to low penetration CCT light. They all have short afterglow life, and lutetium oxysulfate phosphor has longer fluorescence life than lutetium oxysulfide phosphor
Keywords: a general amorphous precursor, lutetium oxysulfate, lutetium oxysulfide, homogeneous precipitation, photoluminescence
Due to its unique physical and chemical properties, rare earth (RE) fluorescent materials have good application prospects in luminescent displays, artificial light sources, biomedical markers, cancer treatment and other fields. Rare earth compounds have good structure, morphology and optical properties in different crystals, and are of great significance in the research field of high-efficiency luminescent materials. There are many kinds of luminescent materials, including rare earth oxide, rare earth oxysulfate, rare earth oxysulfide, rare earth carbonate [1], etc. Among the above materials, rare earth oxysulfate and rare earth oxysulfide have attracted much attention in the field of high energy physics due to their high density, high chemical and thermal stability, high X-ray conversion efficiency and good comprehensive performance. It has great potential and wide application in X-ray intensifying screen, scintillator material, X-ray computed tomography (X-CT) [2], etc. Therefore, it is very important to explore the preparation methods and fluorescence properties of Lu2O2SO4:Eu3+/Lu2O2S:Eu3+ phosphors.
Rare earth oxysulfate (Re2O2SO4) is one of the most important applications in industry as the precursor of Re2O2S synthesis. Usually, Re2O2SO4 is a substance composed of stacked [Re2O2]2+ layers and [SO4]2- layers, while the crystal structure of Re2O2S is typically composed of interlaced [Re2O2]2+ layers and [S]2- layers [3]. Due to the lanthanide contraction principle, the radii of La~Lu atoms gradually decrease with the increase of atomic number, so the atomic radius of lutetium is the smallest, because it is in the last place of lanthanide.Its physical and chemical properties are stable because its 4f electron layer is arrangement reaches its limit. Among them, Re2O2SO4 has chemical and optical properties that other materials do not possess, such as low toxicity, unpaired electrons, long lifespan, etc. In theory, powder materials have various differences in performance due to differences in radius and shape.The powder materials with uniform size and similar shape have higher efficiency [4, 5]. In addition, a large number of studies have shown that the reduction of nanometer size plays an important role in the physical and chemical properties of various nanometer materials.
In the past decade, Ln2O2SO4 powder has been synthesized by many methods due to its excellent properties and broad application prospects. In 2013, Lixin Song prepared europium-doped yttrium oxysulfate (Y2O2SO4:Eu3+) nanoflakes by electrospinning, and then calcined them in a mixture of sulfur dioxide and air at 1000 ℃ [6]. The advantage of electrospinning is the most mild and environmentally friendly, and the shape of the product obtained from oxysulfide is controllable. However, the spinning process is greatly affected by the temperature and humidity of the environment, which will have a certain impact on the shape of the product and its uniformity [7].
In 2014, the above team synthesized Gd2O2SO4:Tb3+ nanosheets in a mixture of SO2 and air through a combination of electrospinning and calcination at 1000 ℃. This type of nanosheet exhibits typical green luminescence [8]. In the same year, our research team synthesized Gd2O2SO4:Eu3+ spherical fluorescent powder through hydrothermal synthesis method, and the prepared Gd2O2SO4 fluorescent powder particles were spherical and well dispersed [9]. The advantage of this method is that the temperature and pressure can be controlled at the same time, and the synthesized products have good morphology and dispersion [10]. In 2015, R. Manigandan reported Gd2O2SO4:Eu3+ nano powder particles by complexation thermal decomposition, and Gd2O2O4 showed strong red photoluminescence [11]. In 2016, our research team synthesized Ln2O2SO4 micro/nano particles through the evolutionary strategy of hydrothermal [12, 13], thermal decomposition and uniform precipitation method based on urea [14]. In 2017, I Aritman. successfully synthesized Gd2O2SO4 submicron powders by the sol gel method with Gd(CH3CO2)3·xH2O, (NH4)2SO4 and CO(NH2)2 as precursors [15]. Moreover, our team prepared La2O2SO4:Tb3+ phosphor by coprecipitation method. And La2O2S:Tb3+ ceramic scintillators were prepared by sintering method under hydrogen atmosphere [16]. In 2019, we synthesized green Gd2O2SO4:Tb3+ phosphor by hydrothermal method [17]. In 2020, R.V. Rodrigues. obtained gadolinium oxysulfate doped with terbium (Gd2O2SO4:Tb3+) by thermal decomposition from sulfate hydrate under dynamic air atmosphere of 1320 to 1400 K [18]. In 2021, our team synthesized a series of Eu3+ doped Lu2O2SO4 phosphors with a diameter of about 800 nm through homogeneous precipitation and calcination at 800 ℃ [19]. The advantage of the homogeneous precipitation method is that it can prepare nanoparticles with adjustable particle size and uniform particle size distribution. The synthesis route is relatively simple, low cost and easy to batch production. It is one of the most promising synthesis technology routes at present [20]. In previous studies, there was no universal precursor for producing both Ln2O2SO4 and Ln2O2S. This time, we propose a simple homogeneous precipitation method to simultaneously prepare general precursors for the production of Lu2O2SO4 and Lu2O2S, and on this basis, we synthesize Lu2O2SO4:Eu3+ and Lu2O2S:Eu3+, respectively. Lu2O2SO4 is synthesized by calcination of the precursor in air. Lu2O2S is synthesized from the precursor, sodium carbonate and sublimed sulfur mixed in a closed crucible and then calcined in air.
Lutetium oxysulfide is a hexagonal structure with a density of 9.02 g·cm-3, which is larger than that of commonly used materials such as Bi4Ge3O12 (7.13 g·cm-3) [20]. Therefore, Lu2O2S has stronger radiation absorption capacity under high-energy radiation [21]. Therefore, the synthesis of rare earth ion doped Lu2O2S scintillation materials with lutetium oxysulfide as the matrix has very significant advantages in the fields of high energy radiation. By consulting the relevant literature, it can be seen that there are few reports on rare earth ion doped lutetium oxysulfide scintillation luminescent materials. Lutetium oxysulfide has strong corrosion resistance and radiation resistance, and the gap between its conduction band and valence band is very wide, making it a good matrix material. The material has great potential and broad application prospects in X-ray sensitization screen, scintillator material, X-ray computed tomography (X-CT) and so on. Therefore, in recent years, the preparation process and characterization technology of Lu2O2S have attracted more and more attention.
In 2007, Jingjing Zhao prepared Lu2O2S:Ce and Y2O2S:Ce powders using oxalate coprecipitation and solid phase coprecipitation methods, and then calcined in a reducing atmosphere of 1150 ℃ [22, 23]. In 2012, Qi Zhao synthesized three-dimensional layered lutetium oxysulfide micro nanostructure through solvothermal methods and subsequent calcination processes. Lu2O2S: Eu3+ powders with flower ball, straw bundle, and flower morphology were obtained by changing the pH value in the reaction system [24]. In 2014, Guowei Wang successfully synthesized a novel three-dimensional nested tetrahedral structure of Lu2O2S:Eu3+ phosphor by using a solvothermal method to prepare the precursor and calcining the precursor in an sulfur atmosphere protected by nitrogen [25]. In 2015, Guowei Wang successfully synthesized nanorod shaped Lu2O2S:Eu3+ phosphors with a diameter of 22 nm and a length of 500 nm by calcining the organic inorganic precursor obtained by solvothermal method at a heating rate of 1 ℃·min-1 in a sulfurized atmosphere under nitrogen protection at 600 ℃ [26]. In 2016, Bowen Zhang successfully prepared Lu2O2S:Eu3+ phosphors with controllable morphology by using a solvothermal method and calcination under a reducing atmosphere to change the pH value [27]. In 2017, Natalie Pasberg synthesized (Lu1−xGdx)2O2S:Tb3+ green phosphors through a combination of urea precipitation and solid phase methods. The results show that the color change of (Lu1−xGdx)2O2S:0.1%Tb3+ is due to the increase in the energy transfer of CTB as the concentration increases [28]. In 2019, Xinxin Xu synthesized two-dimensional sheet shaped Lu2O2S:Eu3+ red phosphors by solid-state method under the protection of hydrogen reduction atmosphere, and focused on the optical properties of lutetium oxysulfide and lutetium oxide [29]. In 2021, our team synthesized a universal amorphous precursor, and Eu3+ doped Lu2O2SO4 and Lu2O2S phosphor was synthesized through a simple coprecipitation method. And Lu2O2SO4:Eu3+ phosphors with similar microscopic morphology were synthesized by calcination method in air, and Lu2O2S:Eu3+ phosphors with similar microscopic morphology in spherical and rod-shaped mixed structures were synthesized by solid-phase method and calcination method under sulfurization atmosphere [30].
Lu2O2SO4 has high absorption capacity for high-energy radiation due to its higher density compared to other lanthanide compounds [31]. In theory, it is a high-quality substrate material, therefore, we should find a simple synthesis method. Homogeneous precipitation is a synthetic technology with relatively simple synthetic route, easy mass production and low operating cost. In this study, we used the modified homogeneous precipitation method to prepare phosphors with lutetium oxide (Lu2O3), sulfuric acid (H2SO4), europium nitrate (Eu(NO3)3·6H2O), ammonia water (NH3·H2O) and urea (CO(NH2)2) as raw materials. Among them, urea provides hydroxyl and carbonate functional groups, and sulfuric acid provides sulfate groups. The structure characteristics, morphology evolution and formation mechanism of the product were discussed by X-ray diffraction (XRD), Field Emission Scanning Electron Microscope (FE-SEM), Thermogravimetric Analysis-Differential Scanning Calorimetry (TG-DSC) and Fourier Transform Infrared Spectroscopy (FT-IR) analysis. And its photoluminescence (PL) was also analyzed.
Synthesis
Using lutetium oxide (Lu2O3, AR), europium nitrate (Eu(NO3)3·6H2O, 99.99%), sulfuric acid (H2SO4, AR), urea (CO(NH2)2, AR), ammonia (NH3·H2O, AR), sodium carbonate (Na2CO3, AR) and sublimated sulfur (S, AR) as the main raw materials, the general precursor of Lu2O2SO4 and Lu2O2S was synthesized by homogeneous precipitation. Among them, Lu2O3 and Eu(NO3)3·6H2O were purchased from Jining Tianyi New Material Co. Ltd, China. Other reagents were purchased from Sinopharm Chemical Reagent Co. Ltd, China.
The first step is to dissolve the corresponding Lu2O3 with sulfuric acid to prepare a Lu2(SO4)3 solution with a Lu3+ ion concentration of 0.02 M. Dissolve Eu(NO3)3·6H2O in deionized water to prepare Eu(NO3)3 solution with a concentration of 0.02 M Eu3+ ions. Next, adjust the pH values of the two solutions to 2.0 using concentrated ammonia water. The mixture was continuously stirred and urea was added at 1:50 molar ratio of Lu3+:CO(NH2)2. Then seal the beaker with aluminum foil and heat it with a water bath, and then the water bath was used for homogeneous precipitation at a temperature of 90 °C and the reaction time of 2 hours. After the water bath was finished and the product in the beaker was cooled naturally to room temperature, and wash them repeatedly with deionized water and anhydrous ethanol in sequence to obtain the precursor. Transfer the precursor to drying oven and dried at 50 ℃ for 12 hours. Then, the dried precursor is transferred to an alumina crucible and calcined in a muffle furnace at 800 ℃ for 2 hours. After cooling, the target product can be obtained. Using the same process to synthesize the target product doped with Eu3+. The doping molar concentrations of Eu3+ ions are 1.5%, 3.0%, 4.5%, 6.0%, 7.5%, and 9%, respectively. The experimental simulation flowchart is shown in Fig. 1(a).
The different precursors obtained were ground in an agate mortar with sulfur and sodium carbonate in the molar ratio of 1:20:5, respectively, where sulfur was used as a sulfur source and sodium carbonate as a flux. The obtained powder was then transferred to a corundum crucible with a mixture of Al2O3 and Al(H2PO4)3 as an adhesive to adhere the lid of the alumina crucible to the bowl, and the well-sealed crucible was placed in a muffle furnace and calcined at 500 ℃ for 2 h, and then cooled with the furnace. The calcined powder was taken out and soaked in 1.5 mol·L-1 dilute hydrochloric acid for 30 min, and then washed repeatedly with deionized water several times to obtain the target Lu2O2S:Eu3+, and the flow chart of experimental simulation is shown in Fig. 1(b).
Characterization
Crystallization behavior and phase composition of the prepared samples were identified by X-ray powder diffraction (XRD) measurements. XRD pattern was performed on a D8 Advance X-ray diffractometer operating at 40 kV and 30 mA with CuKα = 0.15406 nm. The data was collected with 2θ value from 10° to 90°. The morphology of the products were observed by field emission scanning electron microscopy (FE-SEM). The FE-SEM using a Hitachi SU8000 microscope operated at an acceleration voltage of 20 kV. Fourier transform infrared spectra were recorded in the region of 4000-400 cm-1 using an Agilent Cary 660 FT-IR spectrophotometer by the KBr method. Differential scanning calorimetry (DSC) and thermogravimetry (TG) were performed with a heating rate of 5 ℃·min-1 in flowing air atmosphere using simultaneous differential thermal analysis and thermo-gravimetry (SDT 2960). Photoluminescence (PL) spectra were obtained on a Hitachi F-4600 fluorescence spectrophotometer equipped with a 150 W Xenon lamp as the excitation source.
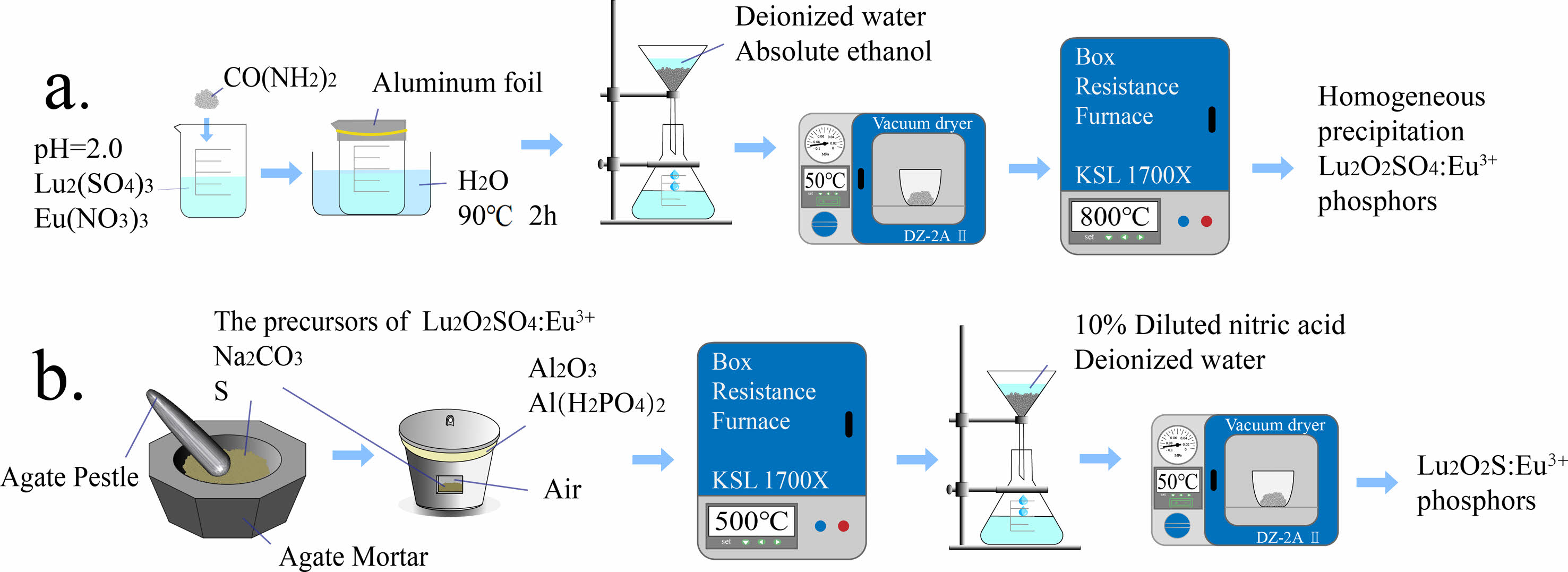
|
Fig. 1 Schematic illustration for the fabrication process of Lu2O2SO4:Eu3+ and Lu2O2S:Eu3+ phosphors. |
Transformation during heating of the precursor
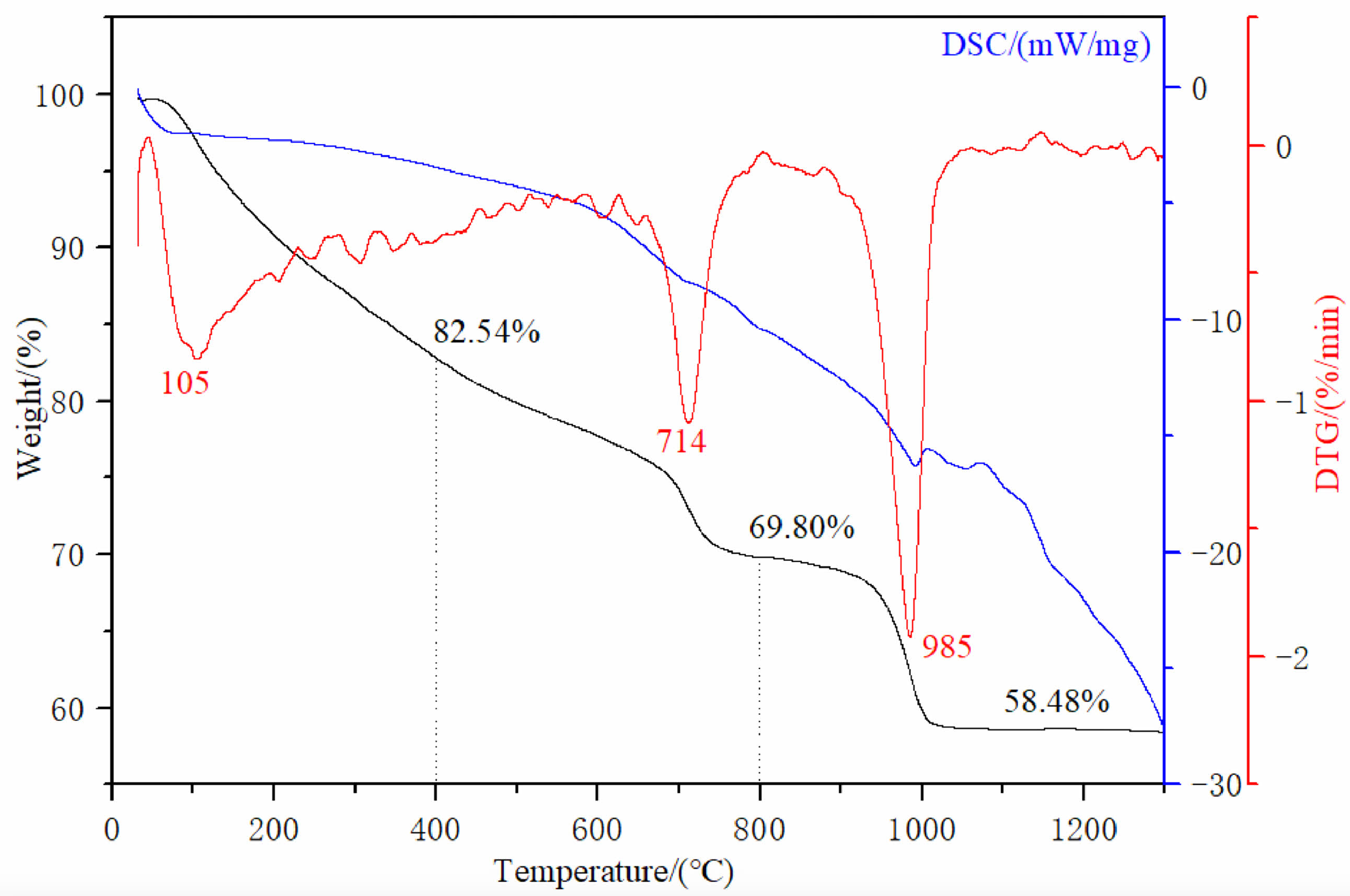
In order to determine the optimal calcination temperature and explore the homogeneous precipitation method to obtain the thermal decomposition transformation of the precursor, DTG-TG-DSC measurements were carried out in the temperature range from room temperature to 1300 ℃ under air atmosphere. The DTG-TG-DSC temperature profiles of the precursor are shown in Fig. 2. In Fig. 2, the blue, black and red curves correspond to the DSC, TG, and DTG curves, respectively.
From Fig. 2, the TG curve shows that weight loss consists of three stages, with a total weight loss rate of approximately 41.52 wt% of the total mass from room temperature to 1300 ℃. The first stage of weight loss was 17.46 wt%, which occurred from room temperature to 400 ℃. On the corresponding DSC curve, around 105 ℃, a corresponding endothermic peak appeared in the figure. A very obvious endothermic peak can be seen on the DTG curve, and a significant weight loss also appeared on the corresponding TG curve. This is due to the weight change caused by the evaporation of water adsorbed on the precursor surface at 100 ℃. When the temperature rises to ~400 ℃, the mass of the precursor decreases to 82.54 wt% of the total mass, which is attributed to the thermal decomposition of the crystalline water in the precursor, and the chemical reaction is shown in Eq. (1):
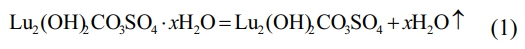
In the second stage, the temperature range is from ~400 ℃ to ~800 ℃, and the corresponding wide endothermic peak appears on the corresponding DSC curve. It can be found on the DTG and TG curves that there is a very obvious sudden weight loss peak at ~714 ℃. When the temperature rises to ~760 ℃, the TG curve becomes flat. At this point, CO32- and hydroxyl groups are removed from the reactant, and the precursor decomposition reaction is completed to form lutetium oxysulfate phase. The mass of the precursor is reduced to 69.80 wt% of the total weight, and the chemical reaction is shown in Eq. (2).
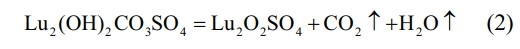
In the third stage, the weight loss temperature range is between ~800 ℃ and ~1300 ℃. A narrow peak centered at ~985 ℃ appears on the DTG curve, accompanied by an obvious endothermic peak on the DSC curve. At this time, the thermal decomposition and desulfurization of lutetium oxysulfate occur to generate lutetium oxide phase, and the mass of the precursor decreases to 58.48 wt% of the total mass. The corresponding chemical reaction formula is shown in Eq. (3). In summary, the optimal calcination temperature for lutetium oxysulfate in this experiment is 800 ℃.
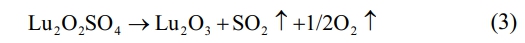
In order to obtain Lu2O2SO4 with high purity and crystallinity, the optimal calcination temperature was determined to be 800 ℃ in this study. The progress of the second and third stage weight loss reactions further confirms that Lu2O2SO4 has a relatively small stable temperature range.
Based on the results of DTG-TG-DSC mentioned above, in order to determine the calcination temperature of the product again, the precursor obtained by homogeneous precipitation method under air atmosphere and the product obtained by calcination at different temperatures (400 ℃, 800 ℃, and 1200 ℃) for 2 hours were detected by XRD patterns.
In Fig. 3(a), the precursor without heat treatment has a certain crystallinity, and there are many obvious diffraction peaks in the diffraction pattern (2θ = 27.43 °, 31.58 °, 35.79 °, 44.15 °, 55.79 °, etc.). However, no corresponding data was found in the JCPDS standard card library to match it, so the precursor is an unknown phase. It is speculated that it may be a new Lu2(OH)2CO3SO4:xH2O phase containing Lu3+ ions, including OH-, CO32-, SO42- groups and crystal water. After the precursor was calcined at 400 ℃ for 2 h in air, as shown in Fig. 3(b), the XRD pattern showed that the diffraction peak became weak and gradually widened, the crystal structure was amorphous, and gradually began to partially crystallize. But no corresponding data was found in the JCPDS standard card library to match it. According to Fig. 3(c), when the calcination temperature is raised to 800 ℃, all diffraction peaks of the sample match well with the data of the Lu2O2SO4 standard JCPDS card (No.00-053-0166), and there are no other impurity peaks. When the calcination temperature is further increased to 1200 ℃ (Fig. 3(e)), all diffraction peaks of the sample can match with the standard JCPDS card (No. 01-086-2475) of Lu2O3. These results are due to the desulfurization process of the product. The above results show that the pure Lu2O2SO4 phase can only be synthesized at 800 ℃, which conforms to the shrinkage law of lanthanide elements. As a result, the temperature of recrystallization and the temperature of dehydroxylation are reduced respectively, so that the stable preparation temperature range of Lu2O2SO4 becomes smaller.
To further investigate the structure of the precursor and the decomposition mechanism of the precursor, we performed FT-IR spectra of the precursor and its calcination products at 400 ℃, 800 ℃, and 1200 ℃, respectively, and the results are shown in Fig. 4.
As shown in Fig. 4(a) that this is the absorption peak of the precursor, including the stretching and bending vibration peaks of OH- (3465 cm-1 and 1654 cm-1), which are the water molecules in the precursor; The asymmetric stretching vibration peaks of C-O (1535 cm-1 and 1404 cm-1) indicate the presence of CO32- ion in the precursor. The S-O antisymmetric stretching vibration peaks (1098 cm-1 and 607 cm-1) indicate the presence of SO42- ion in the precursor.The precursor is mainly Lu2(OH)2CO3SO4 composed of lutetium ion, hydroxyl group, carbonate group and sulfuric acid group, and contains a small amount of crystal water. When the calcination temperature of the precursor was increased to 400 ℃ (Fig. 4(b)), it can be observed that the precursor undergoes high-temperature decomposition and the OH- absorption peak weakens. At the same time, it can be observed that the vibration peak of the carbonate also weakens to a very low level, indicating that carbonate groups and water molecules in the precursor begin to decompose from the precursor as the temperature increases, and other absorption peaks do not show significant changes. When the calcination temperature is further increased to 800 ℃ (Fig. 4(c)), it can be observed that the OH- absorption peak disappears, and the absorption peak of the carbonate group also completely disappears. It can be observed that the SO42- absorption peak splits into a series of sharp absorption peaks (1240, 1135, 1107, 978, 662, and 607 cm-1), but the 978 cm-1 vibration peak of the sulfate group still exists, which indicated that the SO42- group still existed and did not decompose at 800 ℃. This is the precursor that generated the Lu2O2SO4 phase during this calcination process. Fig. 4(d) shows the FT-IR spectrum of the calcined product when the temperature was increased to 1200 ℃. The analysis revealed that all sulfate vibrational peaks disappeared and the vibrational peaks of Lu-O became sharper, indicating that Lu2O2SO4 had gradually transformed into Lu2O3 as the desulfurization progressed, which might further enhance the attraction to O2- and Lu3+ ions. This is in general agreement with the above-mentioned XRD and DTG-TG-DSC results.
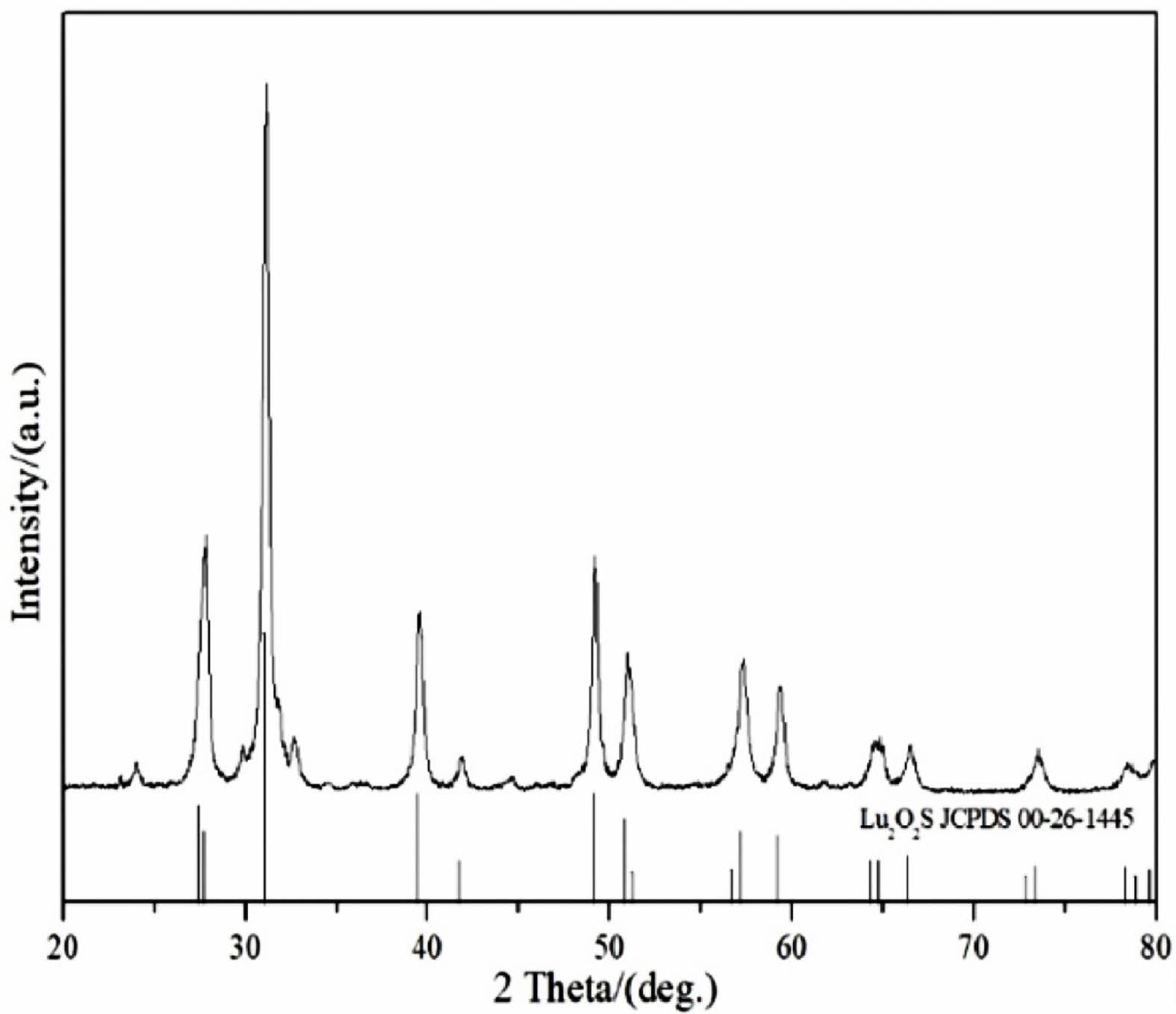
To determine the precursor calcination products under sulphuration atmosphere, Fig. 5 shows the XRD patterns of Lu2O2S. As shown in Fig. 5, the Lu2O2S obtained by homogeneous precipitation method matches well with the JCPDS card (No. 00-26-1445) data of the standard Lu2O2S.
Morphological analysis
Fig. 6 shows the FE-SEM pictures of the precursor and its calcined product Lu2O2SO4:Eu3+ in air atmosphere, and the calcined product Lu2O2S:Eu3+ in sulphuration atmosphere. The SEM photos show that the precursor is generally dispersed, but the size is uniform, and is composed of spherical structure distribution with a diameter of about 1 μm (Fig. 6(a)). However, the obtained Lu2O2SO4 microstructure looks like a spherical submicrosphere shape, the average straight diameter is about 1 μm, which is basically consistent with the shape of the precursor, and has good uniformity (Fig. 6(b)). It is basically consistent with the shape of the precursor and has good uniformity (Fig. 6(b)). When calcined at high temperature, the crystallinity gradually increases, and the crystal agglomerates, and the dispersion becomes poor. Finally, the product Lu2O2S:Eu3+ produced by calcining the precursor under sulphuration atmosphere basically inherits the morphology of the precursor (Fig. 6(c)), which can be found to have higher clarity and better crystallinity.
Photoluminescence properties
By testing found all Lu2O2SO4:x%Eu3+ phosphor with similar PL spectrum curve shape, the excitation spectra, emission spectra and afterglow decay curves of Lu2O2SO4:7.5%Eu3+ phosphor and Lu2O2S:7.5%Eu3+ phosphor are analyzed in this study, respectively. In this paper, the photoluminescence (PL) spectra of Lu2O2SO4:7.5%Eu3+ phosphor and Lu2O2S:7.5%Eu3+ phosphor were recorded at room temperature. Fig. 7 shows the PL excitation and emission spectra of Lu2O2SO4:7.5%Eu3+ and Lu2O2S:7.5%Eu3+ phosphors. As shown in Fig. 7(a), the excitation spectrum of Lu2O2SO4:7.5%Eu3+ obtained by homogeneous precipitation (λem=618 nm) is composed of seven excitation peaks located at 260, 320, 368, 380, 396, 415 and 465 nm. The remaining five peaks are 7F0-5D4, 7F0-5G2-6, 7F0-5L6, 7F0-5D3, and 7F0-5D2 internal structure 4f-4f transitions of Eu3+ ions in the oxysulfate lattice. The emission spectra of Lu2O2SO4:7.5%Eu3+ phosphor are shown in Fig. 7(b), which contains a series of peaks centered at about 581, 620, 655, and 701 nm. They are attributed to the typical Eu3+ ion migration of 5D0-7Fj, which are 5D0-7F1, 5D0-7F2, 5D0-7F3, and 5D0-7F4, respectively.
Among these peaks, the peak at 620 nm is mainly the 5D0-7F2 diffraction of Eu3+ ions, that is the typical red light emission. Interestingly, there is no typical emission peak in the Lu2O3 at 612 nm, indicating the formation of Lu2O2SO4:Eu3+ phosphors. Fig. 7(c,d) is the excitation and emission spectra of Lu2O2S:Eu3+ phosphors.
The excitation spectrum of Lu2O2S:7.5%Eu3+ obtained by homogeneous precipitation (λem = 628 nm) is composed of five excitation peaks located at 270, 325, 368, 392 and 465 nm, the first and second peaks are attributed to the CTB between O2--Eu3+ and S2--Eu3+, respectively, and the remaining three peaks are 7F0-5D4, 7F0-5L6, 7F0-5D2, which are the regional peaks are all internal structural 4f-4f transitions of Eu3+ ions in the oxygen sulfate lattice.The emission spectrum of the Lu2O2S:7.5% Eu3+ phosphor is shown in Fig. 7(d), as it contains some peaks centered at 592, 628, and 709 nm. They are attributed to the typical Eu3+ ion migration of 5D0-7Fj (j = 1, 2, 4), respectively.
Fig. 8 shows the relationship curve between PL strength and Eu3+ ion concentration of Lu2O2SO4:x%Eu3+ and Lu2O2S:x%Eu3+ phosphors. The concentrations of Eu3+ ions were 1.5%, 3.0%, 4.5%, 6.0%, 7.5% and 9.0% respectively. In Fig. 8(a), the luminous intensity increases as the x value increases from 1.5 to 3.0, and then the luminous intensity gradually increases slowly as the x value increases from 3.0 to 7.5. After reaching the maximum value x = 7.5, it can be seen from the above figure that the luminous intensity of the material gradually decreases. As shown in Fig. 8(b), when the x value increases from 1.5 to 3.0, it can be seen that the luminous intensity of the material basically does not change. When the x value increases from 3.0 to 4.5 in the figure, the luminous intensity increases rapidly. When the x value increases to 6.0, it can be seen that the luminous intensity does not continue to increase, but shows a downward trend. When the x value increases to 7.5, it can be seen from the curve that the luminous intensity of the material has increased to the maximum value, and then the luminous intensity gradually decreases. This phenomenon may be caused by "concentration quenching", which may be due to the exchange effect in the luminescence process, which dominates the energy transfer between Eu3+ ions, the energy transfer between Eu3+ activators is related to the critical distance, and due to the existence of single-particle boundary, the long-range interaction of single submicron particles is inhibited to a certain extent. Generally, the light activation will increase with the increase of Eu3+ ions, and the luminous intensity will also increase. However, with the increase of Eu3+ ions, the distance between Eu3+ ions will gradually decrease to the critical distance of Eu3+ ions, resulting in concentration quenching. This also proves to some extent that Lu2O2SO4 has fewer defects than Lu2O2S, and the luminous intensity is higher.
In order to further study the fluorescence characteristics of Lu2O2SO4:7.5%Eu3+ and Lu2O2S:7.5%Eu3+ phosphors, the color emission characteristics were described by calculating parameters such as CIE color coordinates (x, y), the strongest emission peak, and CCT. The data parameters of CIE are summarized in Table 1.
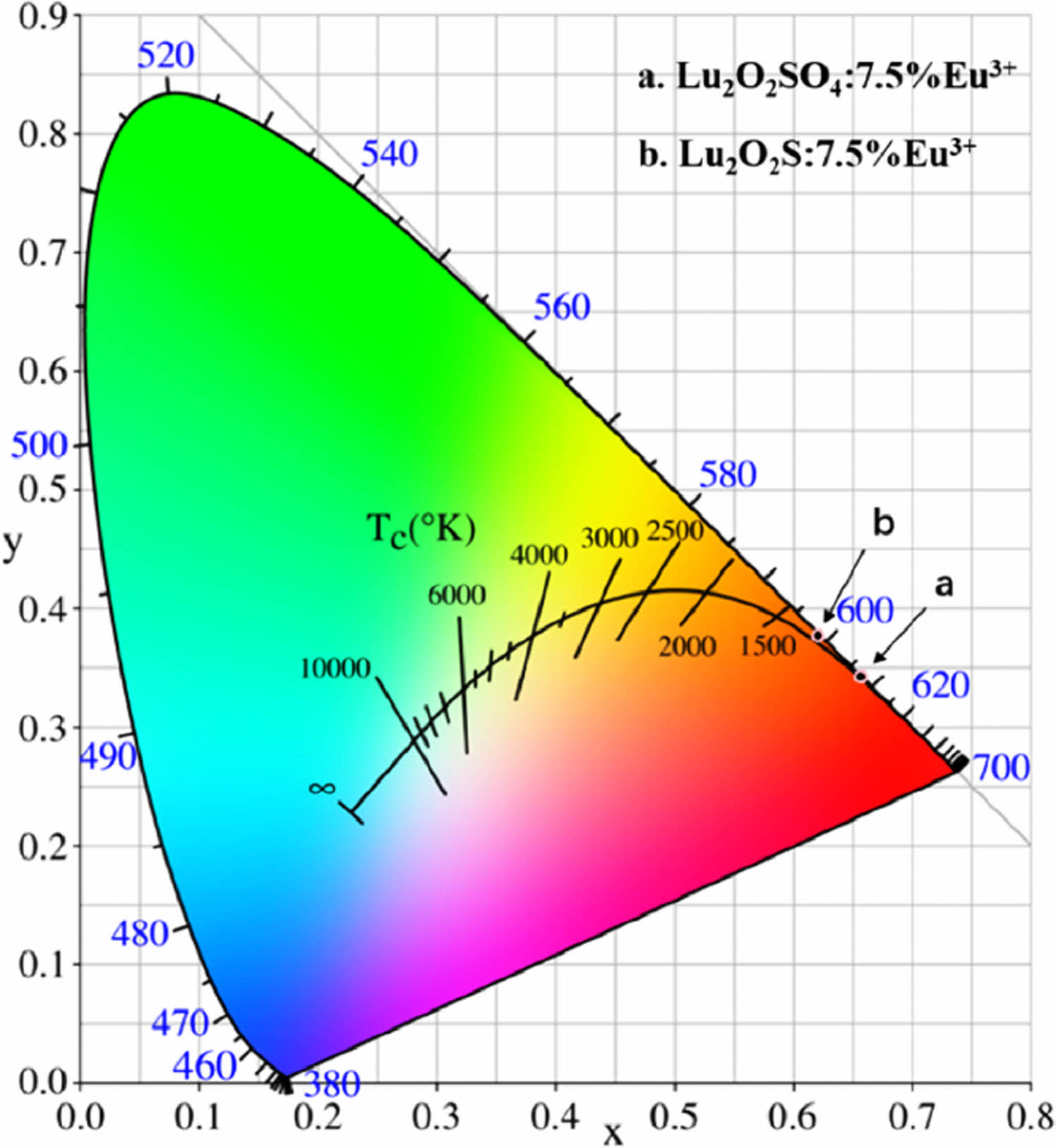
The color coordinates are calculated using color calculation software, and the CIE chromaticity diagram is shown in Fig. 9. CCT is calculated using McCamy's empirical formulas (Equations 4.14 and 4.15).

In Fig. 9, the CIE chromaticity coordinate of Lu2O2SO4: 7.5%Eu3+ is (0.6573, 0.3423), corresponding to red-orange and orange light. In Fig. 9(b), the CIE chromaticity coordinate of Lu2O2S:7.5%Eu3+ is (0.621, 0.3769), corresponding to orange. In addition, their CCT values are 2886 K and 1863 K, which belong to low penetration CCT luminescence and can be used in medical equipment and other fields.
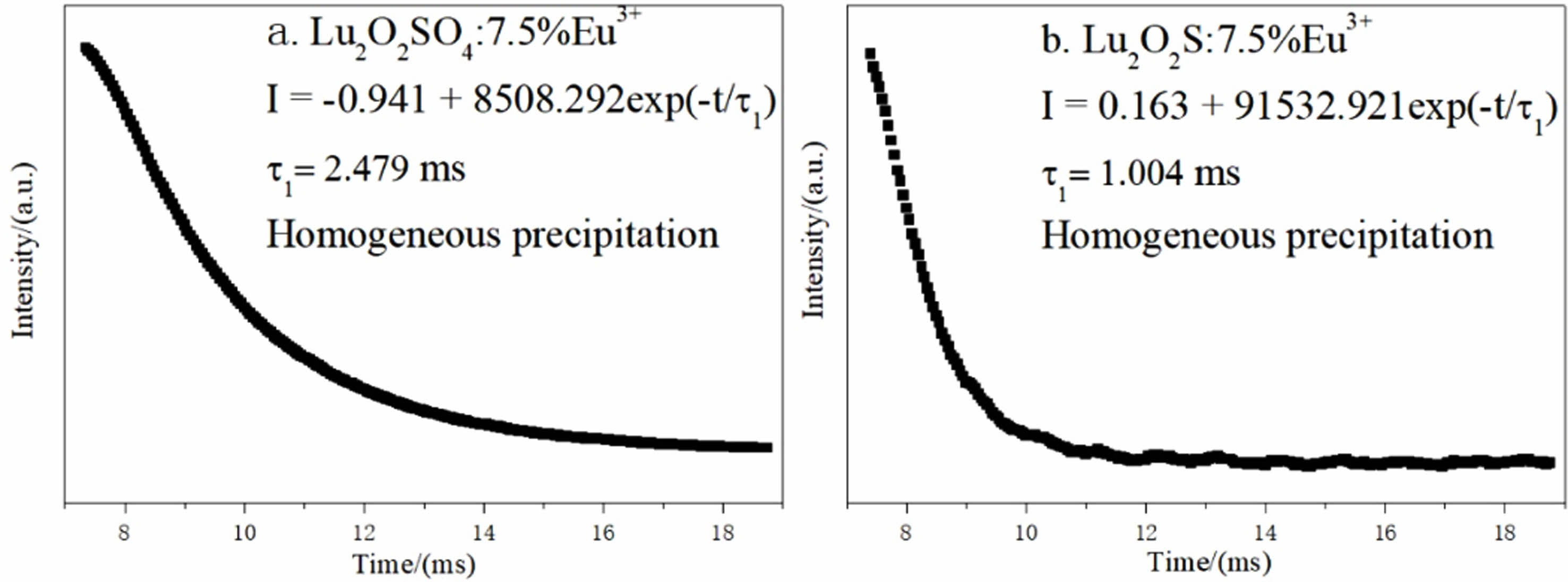
Fig. 10(a) is the afterglow decay curve of Lu2O2SO4: 7.5%Eu3+ phosphor obtained under the excitation of 270 nm ultraviolet light, and Fig. 10(b) is the afterglow decay curve of Lu2O2S:7.5%Eu3+ phosphor under the excitation of 330 nm ultraviolet light, which are monitored at 618 nm and 628 nm. Their attenuation curves can be well fitted with single exponential function (Eq. 6).

Where It and I0 represent the luminous intensity and initial luminous intensity at time t, τ1 is the afterglow luminous lifetime of Eu3+, and A is the fitting constant. The specific single-index fitting formula of the target product obtained in this chapter is shown in Fig. 10.
The fluorescence lifetime of Lu2O2SO4:7.5%Eu3+ and Lu2O2S:7.5%Eu3+ phosphors obtained by homogeneous precipitation method is 2.479 ms and 1.004 ms. They all have short afterglow life, and lutetium oxysulfate phosphor has longer fluorescence life than lutetium oxysulfide phosphor.
|
Fig. 2 DTG-TG-DSC curves of precursor obtained by homogeneous precipitation under air atmosphere. |

|
Fig. 3 XRD patterns of the precursor and its calcined products at different temperatures. |

|
Fig. 4 FT-IR spectra of precursor and its calcination products. |
|
Fig. 5 XRD patterns of Lu2O2S. |

|
Fig. 6 FE-SEM images of (a) precursor, (b) Lu2O2SO4:Eu3+ and (c) Lu2O2S:Eu3+ phosphor. |

|
Fig. 7 L excitation and emission spectra of Lu2O2SO4:7.5%Eu3+ and Lu2O2S:7.5%Eu3+ phosphors. |

|
Fig. 8 Relationship between the Eu3+ molar doping concentration of Lu2O2SO4: x%Eu3+ and Lu2O2S:x%Eu3+ phosphors and their 5 D0- 7 F2 most intense emission light intensity. |
|
Fig. 9 CIE chromaticity diagram of (a) Lu2O2SO4:7.5%Eu3+ phosphors and (b) Lu2O2S:7.5%Eu3+ phosphors. |
|
Fig. 10 Fluorescence decay curves of Lu2O2SO4:7.5%Eu3+ and Lu2O2S:7.5%Eu3+ phosphors corresponding to their 5 D0- 7 F2 strongest transition light. |
|
Table 1 Summary of CIE colorimetric information for: (a) Lu2O2SO4:7.5%Eu3+ by homogeneous precipitation; (b) Lu2O2S:7.5%Eu3+ by homogeneous precipitation. |

A general amorphous precursor composed of Lu3+, SO42-, CO32- and OH- groups was prepared by homo- geneous precipitation method. Eu3+ doped Lu2O2SO4 and Lu2O2S phosphors were synthesized, respectively. Compared with other methods, the main advantage of homogeneous precipitation method is to replace the sulfur source and simplify the experimental steps. On this basis, Lu2O2SO4:Eu3+ phosphor with quasi-spherical structure and Lu2O2S:Eu3+ phosphor with average particle size of about 1 μm were synthesized by calcination in air atmosphere and closed sulphuration atmosphere created by the solid phase method. They all show typical red emission of Eu3+ ions under ultraviolet excitation. The conclusion of fluorescence performance is summarized as follows. The optimum molar doping concentration of Lu2O2SO4:x%Eu3+ and Lu2O2S:x%Eu3+ phosphors obtained by homogeneous precipitation method is 7.5%, and their CIE coordinates are (0.6573, 0.3423) and (0.621, 0.3769), corresponding to red orange and orange luminescence, and the CCT values are 2886 K and 1863 K, which belong to low permeability CCT light, and have good application prospects in medical and other fields. Their fluorescence decay curves can be well fitted with a single exponential function, and the fluorescence lifetime (τ) is 2.479 ms and 1.004 ms, respectively. In addition, under the same concentration of Eu3+ doping, Lu2O2SO4:Eu3+ phosphor with the same near-spherical morphology has stronger emission intensity, higher CCT value and slightly higher fluorescence lifetime than Lu2O2S:Eu3+ phosphor.
This work was supported by the National Natural Science Foundation of China (Grant No. 51802136).
- 1. X.J. Wang, X.J. Wang, Z.H. Wang, Q. Zhu, G. Zhu, C. Wang, S.Y. Xin, and J.G. Li, J. Am. Ceram. Soc. 101[12] (2018) 5477-5486.
-

- 2. X. Li, L.T. Gao, and J.B. Lian, Mater. Tehnol. 56[6] (2022) 669-676.
-
- 3. M. Machida, T. Kawano, M. Eto, D.J. Zhang, and K. Ikeue, Chem. Mater. 15[4] (2007) 954-960.
-
- 4. J.B. Lian, P. Liang, B.X. Wang, and F. Liu, J. Ceram. Process. Res. 15[6] (2014) 383-388.
-
- 5. J.B. Lian, F. Liu, P. Liang, J.M. Ren, and F. Liu, J. Ceram. Process. Res. 17[7] (2016) 752-757.
-
- 6. L.X. Song, X.L. Shao, P.F. Du, H.B. Cao, Q. Hui, T.H. Xing, and J. Xiong, Mater. Res. Bull. 48[11] (2013) 4896-4900.
-
- 7. G.X. Xu, X.T. Sang, J.B. Lian, N.C. Wu, and X. Zhang, Key Eng. Mater. 807 (2019) 1-10.
-
- 8. L.X. Song, P.F. Du, Q.X. Jiang, H.B. Cao, and J. Xiong, J. Lumin. 150 (2014) 50-54.
-
- 9. J.B. Lian, F. Liu, X.J. Wang, and X.D. Sun, Powder Technol. 253 (2014) 187-192.
-
- 10. D.L. Geng, G.G. Li, M.M. Shang, C. Peng, Y. Zhang, Z.Y. Cheng, and J. Lin, Dalton Trans. 41[10] (2012) 3078-3086.
-
- 11. R. Manigandan, K. Giribabu, R. Suresh, S. Munusamy, S. Praveen kumar, S. Muthamizh, T. Dhanasekaran, A. Padmanabana, and V. Narayanan, RSC Adv. 5[10] (2014) 7515-7521.
-
- 12. J.B. Lian, F. Liu, J. Zhang, Y.Y. Yang, X.R. Wang, Z.R. Zhang, and F. Liu, Optik. 127[20] (2016) 8621-8628.
-
- 13. F. Liu, X.R. Wang, Y.Y. Yang, Z.R. Zhang, and J.B. Lian, J. Ceram. Process. Res. 17[20] (2016) 1287-1291. WOS:000397042400015
-
- 14. X. Li, and J.B. Lian, Optik. 127[1] (2016) 401-406.
-
- 15. I. Aritman, S. Yildirim, M.F. Ebeoglugil, M. Yurddaskal, K. Ertekin, and E. Celik, J. Australas. Ceram. Soc. 53 (2017) 457-463.
-
- 16. G.X. Xu, J.B. Lian, N.C. Wu, X. Zhang, and J. He, J. Ceram. Sci. Technol. 9[3] (2018) 345-352.
-
- 17. X.T. Sang, J.B. Lian, N.C. Wu, X. Zhang, and J. He, Polyhedron. 169 (2019) 114-122.
-
- 18. R.V. Rodrigues, L. Marciniak, L.U. Khan, E.J.B. Muri, P.C.M. Cruz, J.R. Matos, W. Strȩk, and A.A.L. Marins, Luminescence. 35[8] (2020) 1254-1263.
-
- 19. M.Y. Liu, J.B. Lian, N.C. Wu, X. Zhang, and J. He, Solid State Sci. 112 (2021) 106520.
-
- 20. K. Okazaki, H. Fukushima, D. Nakauchi, G. Okada, D. Onoda, T. Kato, N. Kawaguchi, and T. Yanagida, Radiat. Meas. 154 (2022) 106773.
-
- 21. L.Q. Liu, E. Ma, R.F. Li, G.K. Liu, and X.Y. Chen, Nanotechnology. 18[1] (2007) 015403.
-
- 22. J.J. Zhao, C.X. Guo, R.W. Guo, and J.T. Hu, J. Alloys Compd. 436[1-2] (2007) 174-177.
-
- 23. J.J. Zhao, C.X. Guo, L.L. Zhang, and J.T. Hu, J. Lumin. 122-123 (2007) 924-926.
-
- 24. Q. Zhao, Y.H. Zheng, N. Guo, Y.C. Jia, H. Qiao, W.Z. Lv, and H.P. You, CrystEngComm. 14 (2012) 6659-6664.
-
- 25. G.W. Wang, H.F. Zou, H.G. Zhang, L.N Gong, Z. Shi, X.C. Xu, and Y. Sheng, Mater. Lett. 128 (2014) 256-258.
-
- 26. G.W. Wang, H.F. Zou, B.W. Zhang, Y.D. Sun, Q.S. Huo, X.C. Xu, and B. Zhou, Opt. Mater. 45 (2015) 131-135.
-
- 27. B.W. Zhang, H.F. Zou, Y.Z. Dai, Y.H. Song, K.Y. Zheng, X.Q. Zhou, and Y. Sheng, RSC Adv. 6[10] (2016) 7846-7853.
-
- 28. N. Pasberg, D. Engelsen, G.R. Fern, P.G. Harris, T.G. Ireland, and J. Silver, Dalton T. 46[24] (2017) 7693-7707.
-
- 29. X.X. Xu, B. Lu, J.X. Hu, and H.B. Chen, J. Lumin. 215 (2019) 116702.
-
- 30. M.Y. Liu, L. Wang, and J.B. Lian, Polyhedron. 210 (2021) 115527.
-
- 31. X. Li, Y.D. Gao, L.Y. Zhang and J.B. Lian, J. Ceram. Process. Res. 23[5] (2022) 709-715.
-
 This Article
This Article
-
2023; 24(4): 640-649
Published on Aug 31, 2023
- 10.36410/jcpr.2023.24.4.640
- Received on Apr 11, 2023
- Revised on Jun 15, 2023
- Accepted on Jul 6, 2023
Services
- Abstract
introduction
experimental procedure
results and discussion
conclusions
- Acknowledgements
- References
- Full Text PDF
Shared
Correspondence to
- Jiao He
-
School of Mechanical Engineering, Liaoning Petrochemical University, Fushun, 113001, P.R. China
Tel : +86-24-56865042 Fax: +86-24-56865042 - E-mail: hejiao@lnpu.edu.cn
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr