





- Effect of pyrolysis atmosphere on preparation of boron carbide from boric acid glycerin
Li Yang, Li Sanxi*, Wang Song, Tian Chengcheng and Otitoju Tunmise Ayode
School of Environmental and Chemical Engineering, Shenyang University of Technology, Liaoning, 110870, China
This article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
The effect of different atmosphere on the particle size and morphology of the B4C was studied by using organic precursor method of low temperature pyrolysis with glycerol borate condensate as raw material. The same mole of boric acid and glycerol were directly mixed, and the condensed product was prepared by dehydration condensation. The precursor powder was pyrolyzed in still air(a), air flow(b), nitrogen flow(c) and argon flow(d) respectively to get rid of excess carbon. The precursors all have a three-dimensional continuous B2O3/carbon network structure, in which the pore size of the carbon network in the static air flow is about 1um, and the pore size of the carbon network in the air flow is about 0.1 um. The influence of the other two gases on the pore size structure of the carbon network is between the static air and the air flow. The morphology and size of B4C powders produced by different synthetic routes (a, b, c, d) were polygonal with 5.7 um, smooth granular with 1.5 um, flake and rod-shaped particles with 14.0 um, and hexagonal with 23.3 um, respectively. This paper is the first time to study the effect of pyrolysis atmosphere on the morphology control of boron carbide from the molecular leve
Keywords: Boron carbide, Different pyrolysis atmosphere, Network structure, Morphology, Glycerol
Boron carbide (B4C) is one of the two known diamond-like carbides [1]. Boron carbide stoichiometry, which determines its properties, demonstrates a wide phase homogeneity range from B4C to B10C. Boron carbide, besides its extreme hardness (up to 35 GPa), has other very interesting properties, such as: low density (2.52 g/cm3), high thermal conductivity (20-30 W/(mK) 20-2400 K), good chemical resistance, and a very high neutron capture cross-section (755 b). Due to its wide range of properties boron carbide is used as an abrasive in polishing and lapping applications, as ballistic armour, and as an absorbent for neutron radiation [1-5]. Due to the high neutron capture cross-section, boron carbide nanopowders can be a potential boron carrier applicable in boron neutron capture therapy (BNCT) [6, 7]. High concentrations of 10B isotope, reaching 20%, which are desirable in radio- therapy, are observed in boron carbide [1, 2, 5]. Boron carbide powders are usually synthesized by boron oxide carbothermic and magnesiothermic reduction, performed at high temperature (1900-2000 °C), and their product requires grinding and purification [7]. The reaction is represented as:
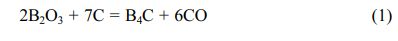
This process is suitable for quantity synthesis because the starting materials are inexpensive and nonhazardous. On the other hand, this process has some disadvantages such as boron loss by B2O3 volatilization and the coarsening of the grains produced, which are caused by the high synthesis temperature. A low-temperature synthetic route of crystalline B4C powder by carbo- thermal reduction using organic precursors including borate ester (B-O-C) bonds prepared from boric acid (H3BO3) and polyols, e.g., cellulose and glucose [8], citric acid [9-11], glycerin [12], phenolic resin [13], and poly(vinyl alcohol) (PVA) [14], has been investigated. The formation of B-O-C bonds in a gel precursor using the sol-gel process enables more homo- geneous dispersion of H3BO3 and polyol as the boron and carbon sources, respectively, thus, the synthesis at a lower temperature of approximately 1500 oC can be achieved. The stoichiometric reaction between hydroxyl groups of H3BO3 and that of a polyol induces sufficient B-O-C bond formation.
Unfortunately, this advantage leads to a large amount of residual free carbon in the obtained product synthe- sized at low temperatures owing to excess carbon compared with that required for carbothermal reduction [Eq. (1)]. This is the greatest disadvantage of the synthesis of B4C powder using an organic precursor. In an attempt to solve the above problem, in previous study, a precursor consisting of B2O3 and amorphous carbon with a stoichiometric composition ratio of carbothermal reduction [Eq. (1)] was prepared by the thermal decomposition of a condensed product pre- pared from H3BO3 and polyols in air [15-18].
The thermal decomposition in air to prepare a homogeneous precursor by removing excess carbon for carbothermal reduction, not by adding excess H3BO3 to condensation. Its synthesis temperature and holding time are lower and shorter with the use of a precursor with a homogeneously arranged B2O3/carbon structure at the nanometer scale [16-18]. It is suggested that the synthesis of B4C powder is affected by the differences in the homogeneity and dispersibility of the B2O3 and carbon components in the precursor. Glycerin, which is a typical polyol, is conveniently used for the preparation of a condensed product with H3BO3. Furthermore, glycerin is an organic solvent of low molecular weight, thus, homogeneous and easy blending is possible with an organic compound [19]. In previous reporter showed that a precursor prepared from the condensed H3BO3-glycerin product by thermal decomposition in air forms a characteristic three-dimensional bicontinuous structure composed of B2O3 and carbon components [16, 25].
In the process of synthesizing boron carbide powder, previous researchers have made great contributions to controlling its morphology and size at the molecular level, but they have not considered the influence of pyrolysis atmospheres on the preparation of boron carbide. Therefore, we attempted to further develop the precursor structure by the difference in pyrolysis atmosphere, to obtain a precursor with a more homo- genously and finely dispersed B2O3/carbon structure derived from the condensed H3BO3-glycerin product. Homogeneous boron carbide is obtained by accurately controlling the atmosphere, temperature and synthesis time. This paper will provide some reference for future researchers in controlling the morphology of boron carbide at the molecular level.
Materials
Boric acid (BA) (99.9%) purchased from Bor Mining Chemical Company, Russia. Glycerin (Gl) (99.0%) purchased from Sinopharm Chemical Reagent Co., Ltd, Shanghai, China. These materials were used as received without any prior treatment in this work.
Synthesis of B4C powder
A condensed product was prepared by dehydration at 150 °C after directly mixing equimolar a mounts of H3BO3 and glycerin. The resulting product was a transparent glassy solid. This condensed product was placed in an alumina crucible and heated in different atmosphere (still air, air flow, N2 flow and Ar flow) at 250 °C for 2 h then at 350 °C for 2 h, which was followed by pyrolysis in air at 500-800 °C for 2.5-5.5 h to obtain a precursor powder from which the excess carbon had been eliminated. The above steps were consecutively performed using a temperature programmed muffle furnace. The obtained black precursor powder was placed in a graphite boat after grinding with an agate mortar and heated at 1300-1500 °C for 1-3 h in an Ar flow (200 ml/min) at a heating rate of 10 °C/min.
Characterization methods
A Shimadzu IR prestige-21 Fourier transform infrared spectrophotometer was used to identify the chemical bonds of the formed precursor from the solid-solid-liquid reaction of (BA-GL). A Rigaku (RAAD-C) x-ray diffractometer operated at 40 kV and 30 mA with monoChromatized CuKα radiation was used to recognize the phases in the final products. AHitachi-SU8010 ultra-high-resolution scanning electron micro- scope (UHRFESEM) operated at 20.0 kV was used to study the morphology of the final product. B2O3 content in the precursor powder was determined as H3BO3 by the titration of mannitol-H3BO3 complex with sodium hydroxide solution using phenolphthalein as an indicator. The B2O3 component was recovered from the precursor powder by washing in hot water.
Boric acid conversion
The conversion rate of boric acid is obtained by the acid value of the remaining boric acid in the product and the initial acid value of the reactant boric acid. Because of the special properties of the product ester and boric acid, the acid value is measured by titration by steps.The specific method was to take 0.5 g (accurate to 0.0001 g) product ester and dissolve it in 25 mL anhydrous ethanol. Using phenolphthalein as indicator, the end point was 1, which was titrated with a certain concentration of NaOH ethanol solution until the white turned pink. The volume of NaOH V1 was recorded.After the titration of the first step, 5 g glycerol was added, and the titration was continued until the second titration turned pink, which was the end point 2. The volume of NaOH V2 was recorded. Calculate with Eq. (1).
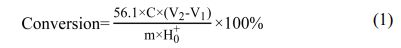
Wherein: H0+-- the initial acid value of boric acid of the reactant, C -- NaOH ethanol solution concentration, M -- the mass of the product ester.
Determination of boron oxide content and C/B2O3
B2O3 in the pyrolysis product will react to generate H3BO3 after absorbing water. A little mannitol is added to enhance the degree of dissociation of boric acid. Then, the content of B2O3 can be obtained according to the titration result by titration with NaOH standard solution and phenolphthalein as indicator.Weighed a certain amount of cracking product powder M1, dissolved it in hot distilled water at 80 ℃, stirred it for 5 min, then filtered and washed it, and the transparent filtrate obtained was H3BO3 solution. The H3BO3 solution obtained after washing was transferred to a conical flask, and 5 drops of mannitol was added to enhance the disintegration of H3BO3. Phenolphthalein was used as an indicator, and then the standard solution of NaOH with a certain concentration was titrated until the solution turned pink, which was the end point of titration. The volume of NaOH was recorded as V.After washing, the pyrolysis product was dried to a constant weight, and the recorded mass was m2. Calculations were made using Eqs. (2 and 3).
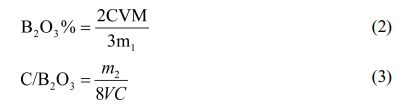
B2O3%—Boron oxide content, M—Relative molecular weight of boron oxide, C/B2O3—Molar ratio of carbon to boron oxide, C—The concentration of an aqueous solution of NaOH.
Structure of condensed products
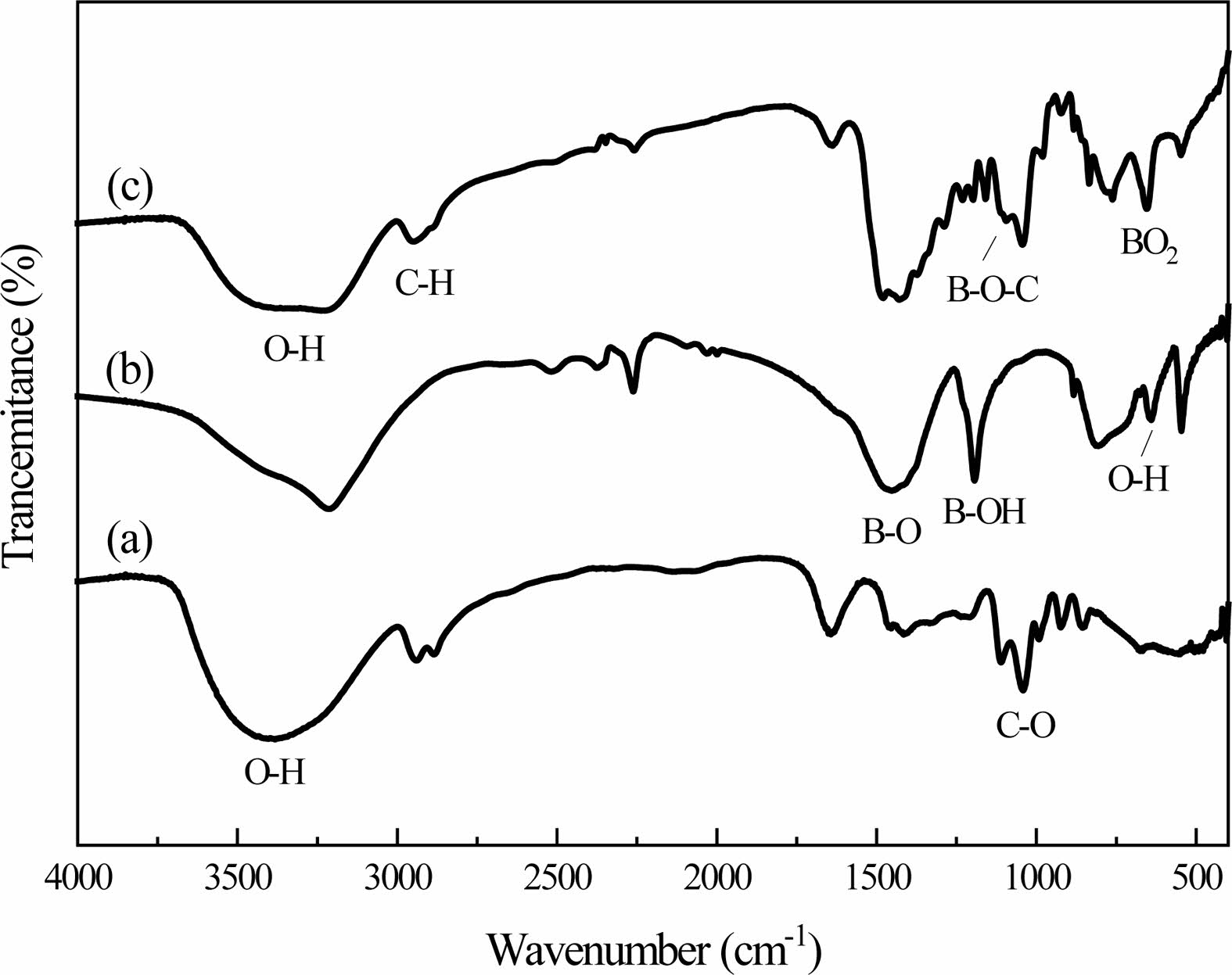
The dehydration condensation reaction is induced by heating a mixture of glycerin dissolved in BA at 150 oC. FT-IR measurement was performed to evaluate the bonding state of the condensed products. The FT-IR spectra of the raw materials and the condensed products are shown in Fig. 1. The absorption peaks assigned to the C-O stretching mode at 987 cm-1 [17], the C-H stretching mode at 2913 cm-1 and the O-H stretching mode at 3000-3500 cm-1 [18] with a wide peak were observed in Fig. 1(a). These peaks are the characteristic absorption peaks of glycerin in the raw material.
The absorption peaks assigned to the BO3 torsion bands at 642 cm-1 with Sharp peak, the O-H torsion bands at 785 cm-1 with Strong and broad peak, B-OH stretching mode at 1189 cm-1 and the B-O stretching mode at 1454 cm-1 [19] were observed in Fig. 1(b). These are the characteristic absorption peaks of boric acid. Fig. 1(c) shows the condensed products that the stretching vibration peak of the B-O-C bond appears at the 1050 cm-1, which is due to the reaction of boric acid and glycerol, which forms a new chemical bond.
However, compared with boric acid, the absorption peak intensity of BO3 torsion bands and the B-O-H stretching mode at 785 cm-1 in the condensed products becomes weaker. Compared the absorption peak inten- sities of these sets of infrared spectra, the characteristic absorption peaks in boric acid and glycerol and the characteristic absorption peaks in condensed products have changed in peak shape and peak position. The weakening of the B-OH bond and the formation of the B-O-C bond in the condensed products both confirmed the occurrence of the esterification reaction.
Conditions for thermal decomposition of condensed products and structure of prepared precursors
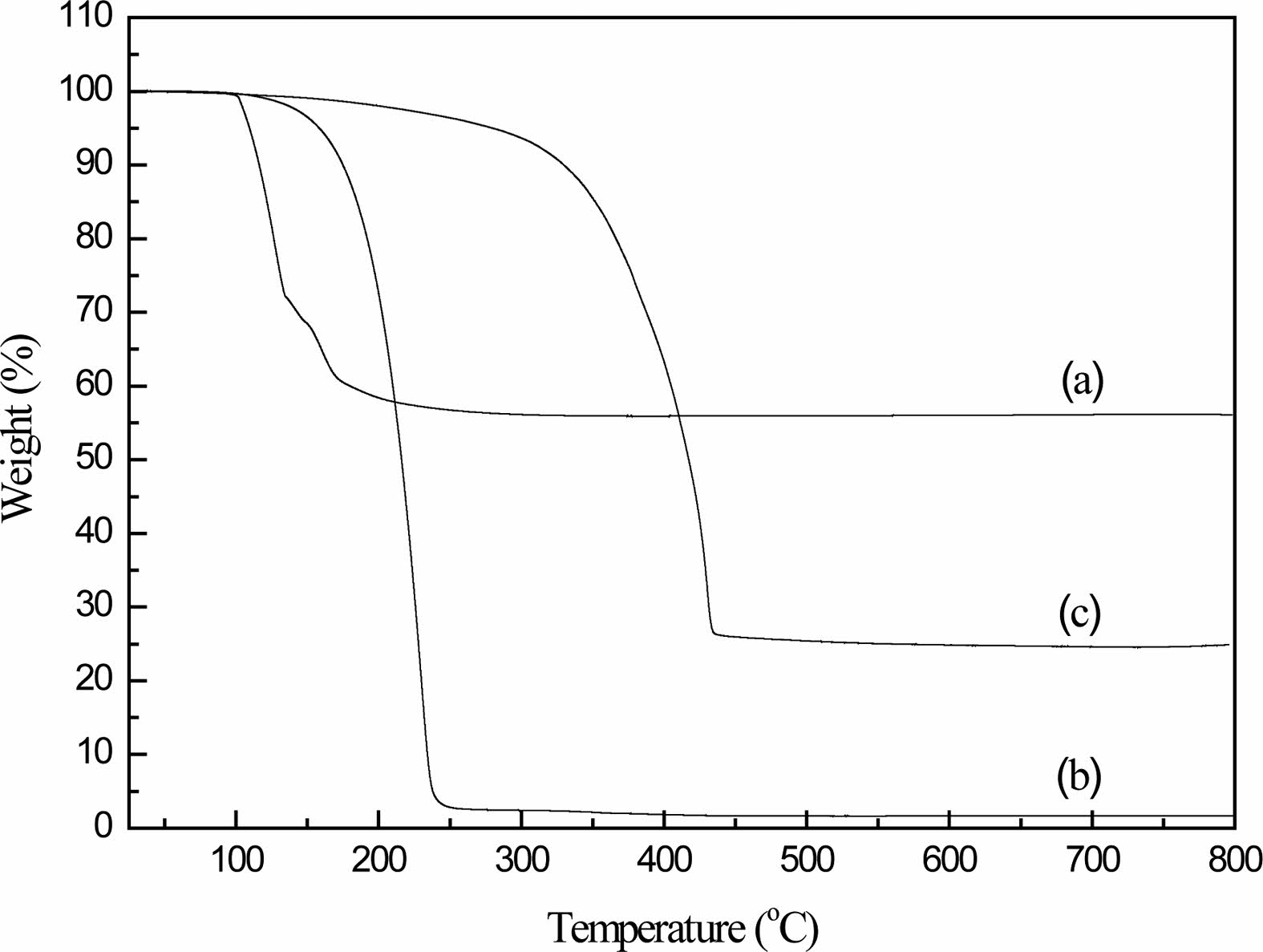
The thermal behavior of the condensed products was investigated. Fig. 2 shows the TG curves of the starting materials and condensed products. The dry boric acid powder begins to thermally decompose at about 100 ℃, and the mass remains unchanged after 200 ℃, with a mass residual rate of 56.1%; after glycerin at 240 ℃, the mass residual rate is only about 1.6% and remains unchanged. It can be judged that the thermal decomposition of boric acid and glycerin is complete after 250 ℃.Within 0-800 ℃, the organic precursor undergoes two thermal decompositions. The first thermal decomposition occurs at 150-300 ℃, and the weight loss is very small at about 10%, which is caused by the disappearance of free water contained in the organic precursor.
The second stage of weight loss occurs at 300-450 ℃. When the decomposition temperature is 350 ℃, the thermal decomposition rate of the organic precursor is the fastest. This is due to the rupture of the eight-membered ring and the carbon skeleton of the organic precursor at a certain temperature [20, 21].
When the temperature is 450-800 ℃, the mass residual ratio remains almost unchanged, indicating that the cracking reaction of the organic precursor occurs at a temperature lower than 450 ℃. It can be known from the chemical equation 1.1 that, according to the stoichiometric ratio, when 1 mol of B4C is produced, the molar ratio of C and B2O3 is 3.5:1, but when the equivalent molar ratio of glycerol and boric acid is added, it is converted into the moles of C and B2O3 The ratio is 6:1, so carbide is excessive at this time.
In order to reduce the molar ratio of C and B2O3 in the carbothermic reduction reaction raw materials, in this experiment, the organic precursor was heated from room temperature to first in different cracking atmospheres (still air, air flow, nitrogen flow and argon flow). 250 ℃, constant temperature heat treatment at this temperature for 2 h, the purpose is to remove the free water contained in the precursor, to ensure that the esterification reaction proceeds to the right; then heat up from 250 ℃ to 350 ℃, heat treatment at this temperature for 2 h, Carry out the preliminary thermal decomposition reaction; and then raise the temperature to the pyrolysis temperature of 500-800 ℃ respectively. After holding at this temperature for 2 h, the pyrolysis product is prepared and used for the later carbothermal reduction to prepare B4C.
Preparation of condensation products
In the process of reaction between boric acid and glycerol, when the reaction exceeds a certain temperature, glycerol is easy to decompose into acrolein to make the finished product yellow, and increase the irritation [16]. Therefore, the control reaction temperature is the key to making qualified finished products. Different material ratio and reaction time will affect the chemical balance of esterification reaction process [22, 26]. On the basis of these theories and experimental influencing factors, the optimal process conditions for the preparation of organic precursors were determined by changing the molar ratio of boric acid and glycerol, reaction temperature and reaction time with the conversion rate of boric acid as the measuring standard.
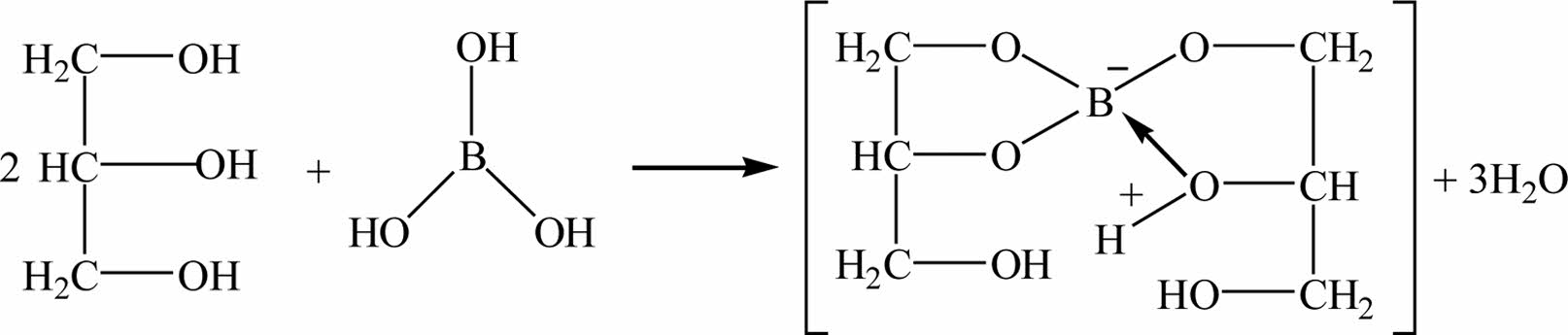
Fig. 3 shows an equation of boric acid and glycerol reaction mechanism. Since H3BO3 is an electron-lacking compound and easy to hydrolyze, it usually exists in the form of monohydrate boric acid H2O·B(OH)3. In the reaction with polyols, B(OH)4+ and H+ are ionized. B(OH)4+ and polyhydroxyl alcohols undergo esterification reaction to remove water molecules and generate a stable complex containing an eight-member ring [23].
The structural formula of glycerol is CH2(OH)- CHOHCH2(OH), which is an organic compound containing three hydroxyl groups. Because the hydroxyl group is a strong hydrophile group, glycerol and boric acid are directly mixed at low temperature to produce dehydration condensation reaction between the hydroxyl groups to generate organic matter containing boron and carbon (glycerol borate). The borate glycerol ester can be used as an organic precursor for the preparation of boron carbide. Fig. 3 shows that the formation of B-O-C bond and the disappearance of B-OH bond are the important basis for the esterification reaction of the reactants.
Effect of molar ratio of reactants on synthesis of organic precursors
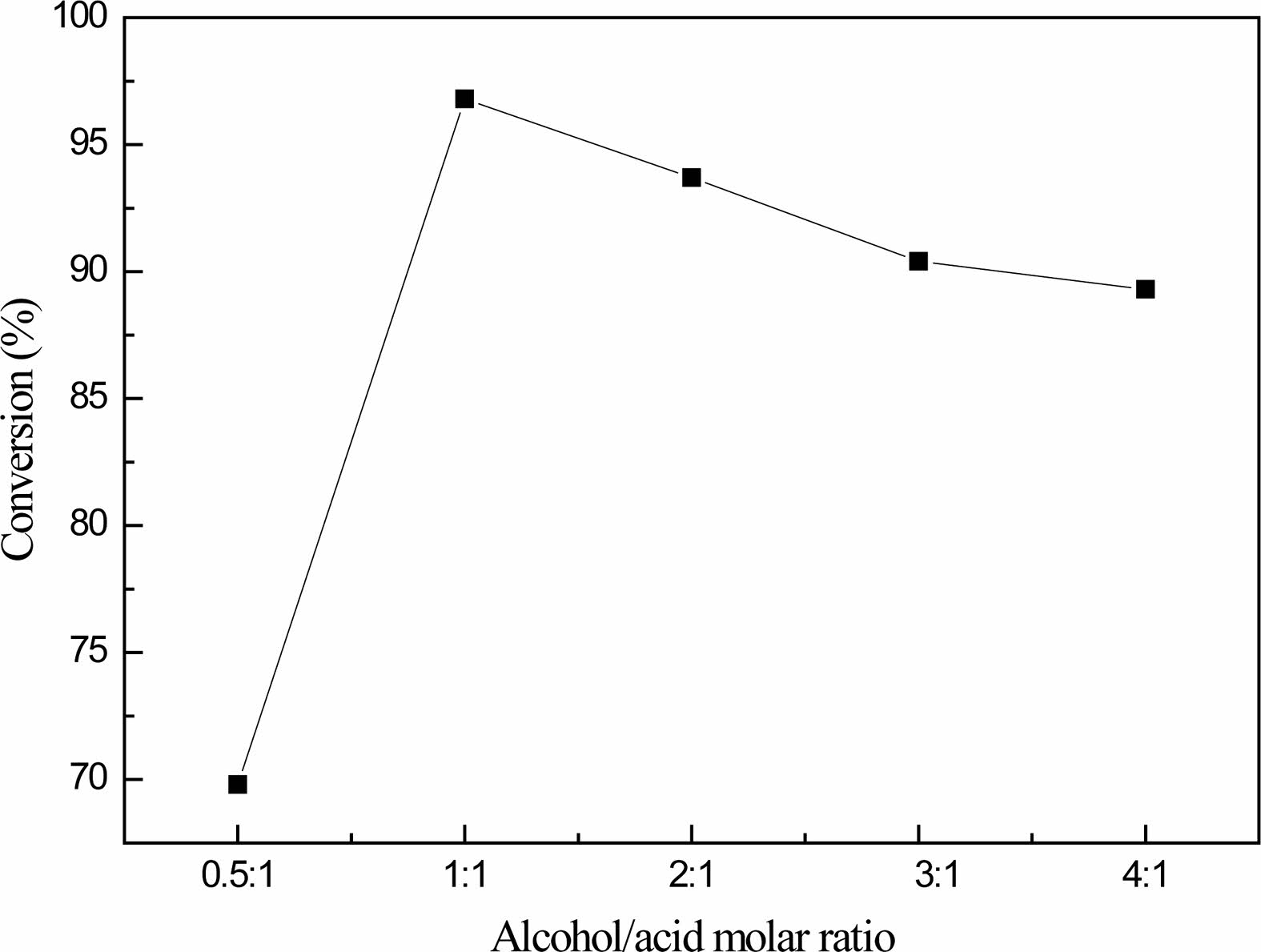
Fig. 4 shows that when the molar ratio of glycerol and boric acid is 0.5:1, the conversion rate of boric acid is 69.8%; when the molar ratio of glycerol and boric acid is adjusted to 1:1, the conversion rate of boric acid is 96.8% at the highest. When the molar ratios of glycerol and boric acid were 2:1, 3:1 and 4:1, the corresponding boric acid conversion rates were 93.7%, 90.4% and 89.3%, respectively. The boric acid conversion rate first increased and then decreased with the increase of the amount of glycerol.
When the amount of glycerol is small, the boric acid is not completely reacted, and finally metaboric acid is formed, which makes the conversion rate of boric acid is not high. When the amount of glycerol is too large, the concentration of boric acid decreases, the reaction is inhibited to the positive reaction, and the conversion rate of boric acid decreases gradually.
When the molar ratio of glycerol to boric acid is 1:1, it is the best molar ratio of reactants to prepare organic precursors. Because there is still a large amount of unreacted boric acid in the product after reaction, in industrial production, the method of cyclic reaction can be used to improve the yield and reduce the waste of boric acid.
Effect of reaction temperature on synthesis of organic precursors
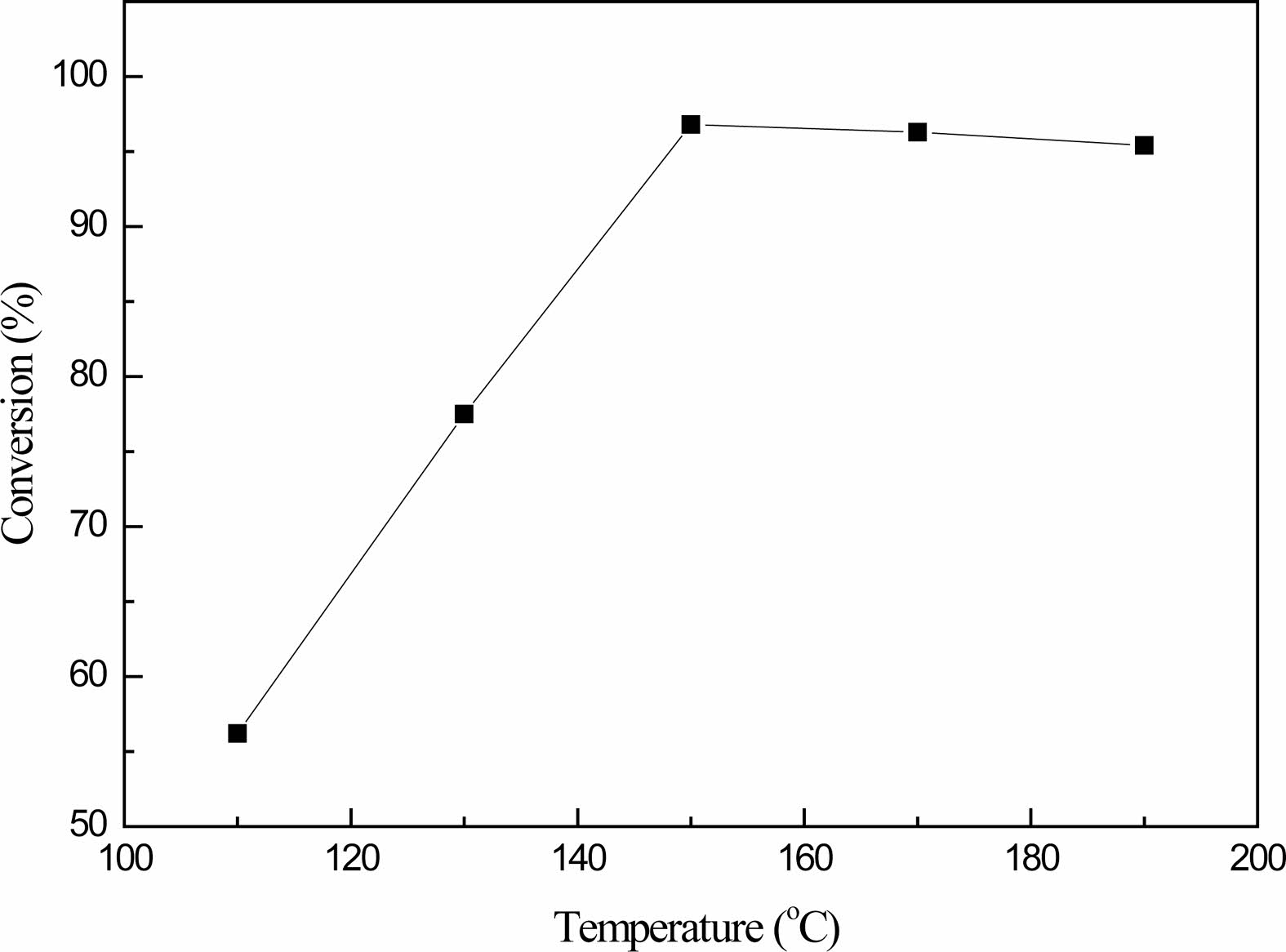
Fig. 5 shows that the reaction temperature has a great influence on the conversion of boric acid. At the beginning, as the temperature increases, the conversion of boric acid increases significantly, and then tends to remain unchanged.At 130 ℃, the conversion rate is only 77.5%. When the temperature reaches 150 ℃, the conversion rate is as high as 96.8%, while the conversion rate drops slightly in the temperature range of 160-190 ℃.Therefore, the higher the reaction temperature, the more promoting the esterification reaction, but from the experimental observation, when the temperature reached 160 ℃, the generated products appeared yellow, with the rise of the reaction temperature, the yellow deepened, this is because the incomplete glycerol, after a certain temperature, generated acrolein; On the other hand, according to the chemical properties of boric acid, when the temperature is too high, the side reaction of boric acid to metaboric acid is intensified and the viscosity of the product is increased.Therefore, the optimal reaction temperature is 150 ℃.
Effect of reaction time on synthesis of organic precursors
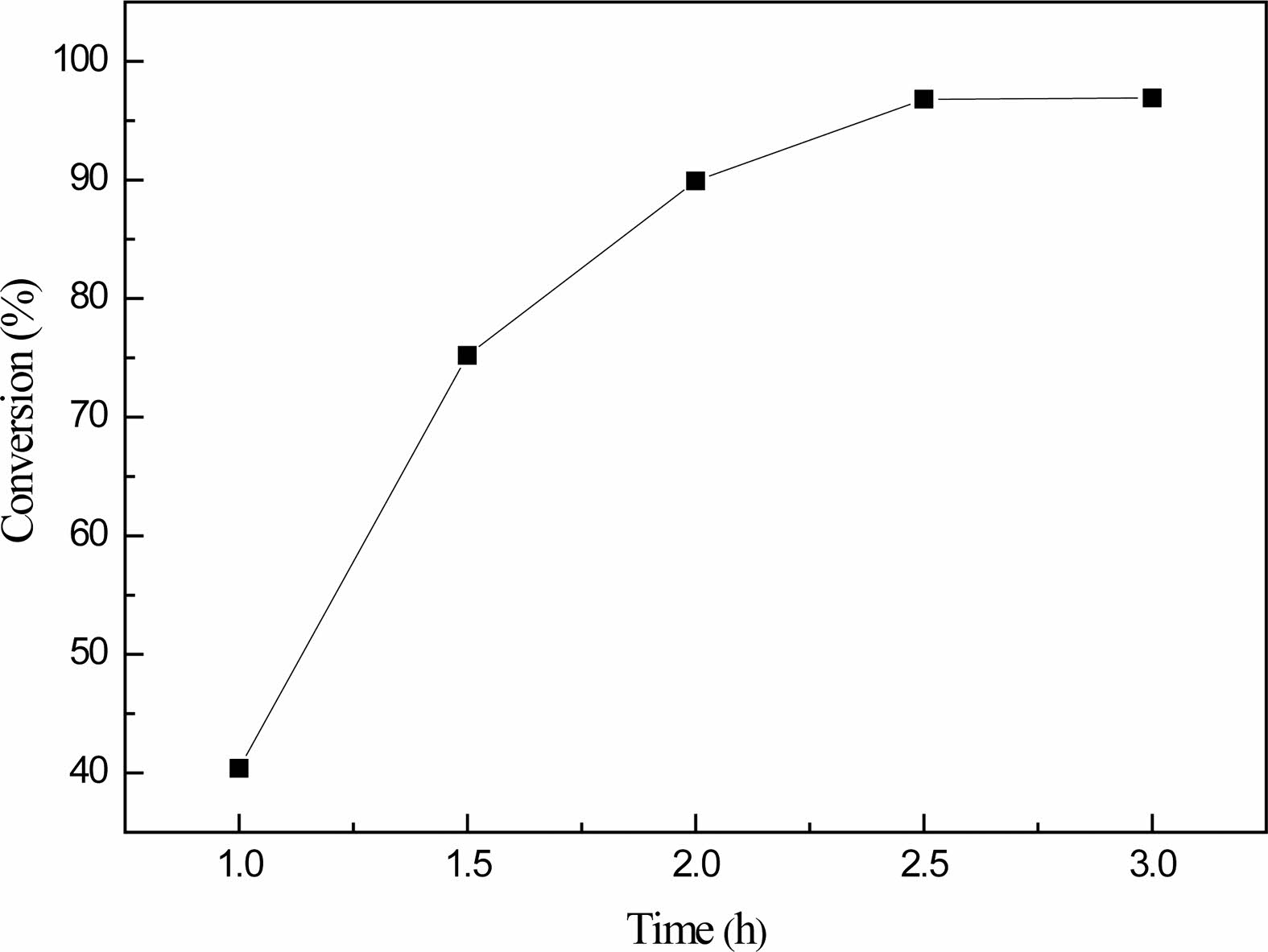
Fig. 6 reflects the influence of different reaction time on the boric acid conversion rate, when the reaction time of 1 h, boric acid conversion rate is only 40.4%, and with the increase of reaction time, the conversion rate of boric acid followed gradually increases, when the reaction time is 1.5 h, boric acid conversion rate increase to 75.2%, when the reaction time increased to 2 h, The conversion of boric acid is close to 90%.The experimental phenomenon shows that there is still a large amount of water vapor from the container volatilization, continue to extend the reaction time to 2.5 h, at this time, the conversion of boric acid up to 96.8%, when the reaction time continues to extend 0.5 h as a step, the conversion of boric acid almost maintained at about 97%, so the best reaction time is 2.5 h.The results showed that when the molar ratio of glycerol to boric acid was 1:1, the reaction temperature was 150 ℃ and the reaction time was 2.5 h, which were the best conditions for the preparation of organic precursors. In the following analysis and experiments, the organic precursors prepared under such process conditions were selected as the raw materials for analysis samples and pyrolysis reactions.
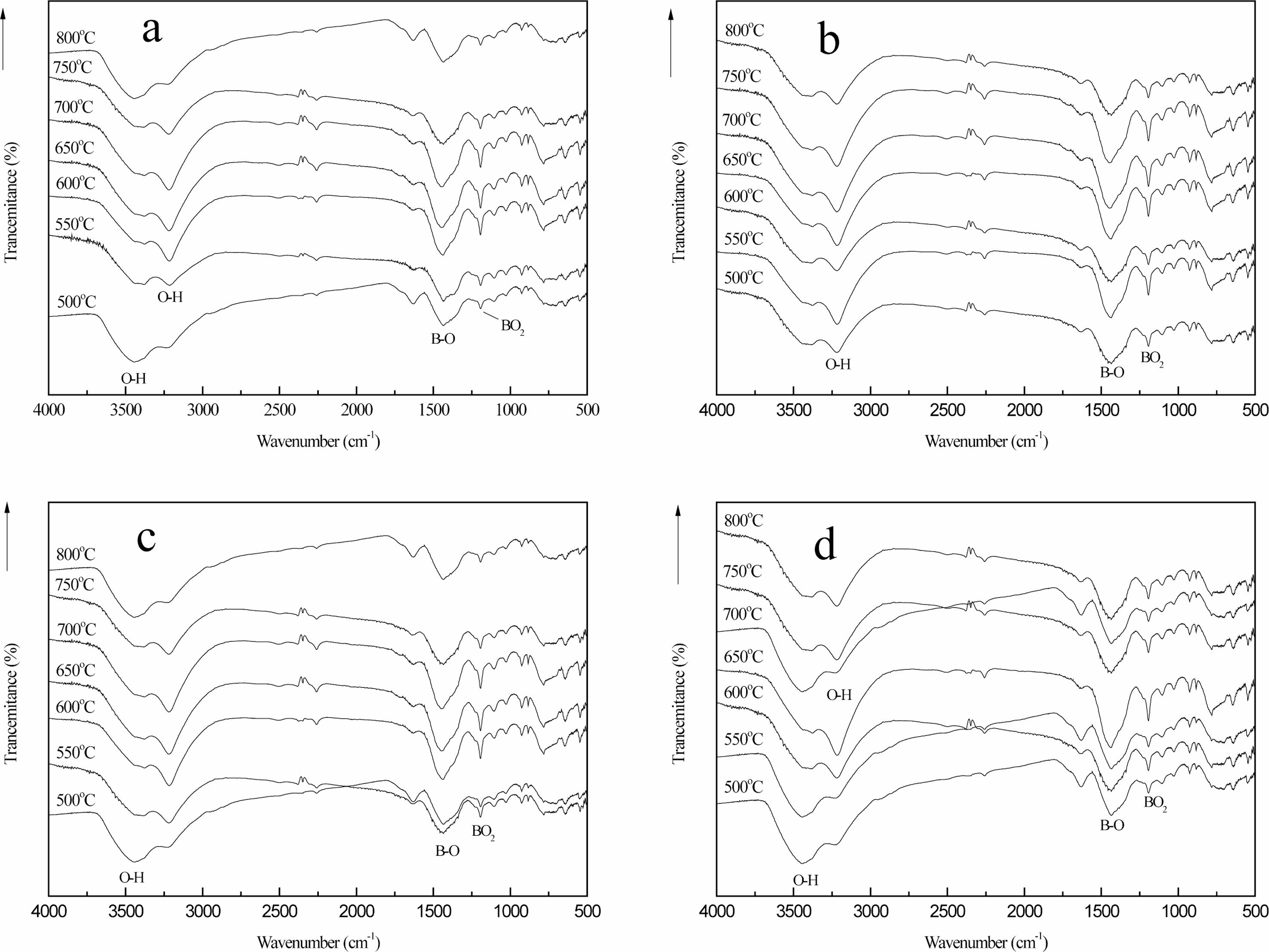
Structural analysis of pyrolysis products
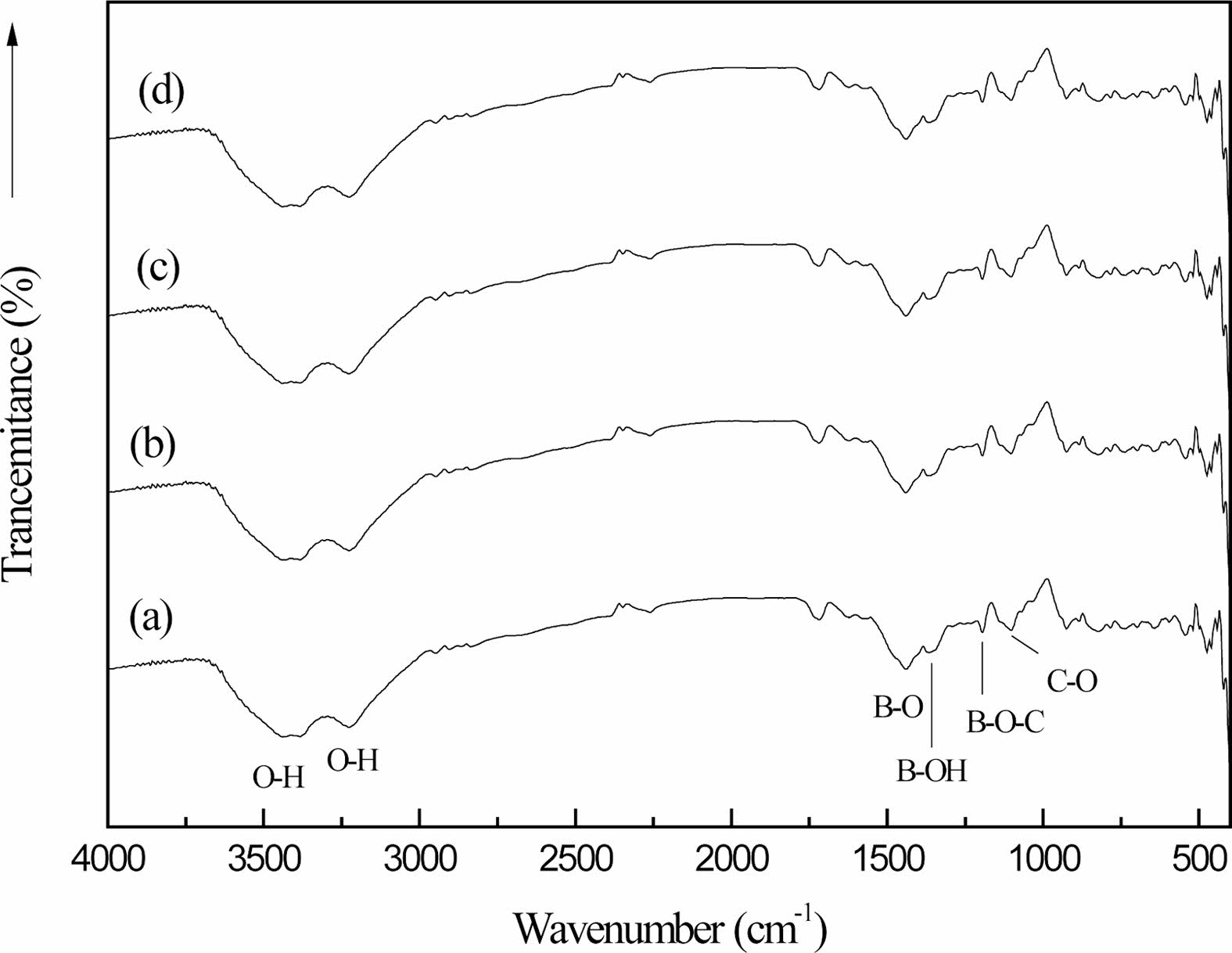
Fig. 7 shows the organic precursors in different cracking atmospheres (static air (a), air flow (b), nitrogen flow (c) and argon flow (d)) after 2 h heat treatment at 250 ℃. Fig. 7 is compared with Fig. 1(c) shows the strength of the stretching vibration peak of the B-O-C bond at 1050 cm-1 is higher than that of the absorption peak in the organic precursors after heat treatment at 250 ℃ for 2 h under different cracking atmospheres. The peak surface is very wide at 3000-3500 cm-1, and the stretching vibration peak of the O-H bond with high strength changes in the peak position and the peak shape, which is transformed into the stretching vibration peak of the O-H bond at 3342 cm-1 and the bending vibration peak of the O-H bond at 3196 cm-1 [27], and the strength of the peak becomes weaker.
The chemical bonds of the organic precursors in the static air, the air flow, the nitrogen flow and the argon flow have not disappeared or changed obviously after the heat treatment at 250 ℃ for 2 h. It is proved that the organic precursors have not undergone obvious chemical changes after the heat treatment at 250 ℃ for 2 h. However, after heat treatment, the O-H bond has obvious displacement changes [2, 30]. There is a large amount of free water volatilization in the organic precursors.
By comparing curves (a), (b), (c) and (d), it can be seen that the vibration peak intensity of B-O-C bond in (b), (c) and (d) is much larger than that in (a). On the other hand, the strength of O-H bond stretching vibration peaks in (b), (c) and (d) is much smaller than that in (a). This is because there is a large amount of free water volatilization in the organic precursor at 250 ℃, and the rate of free water volatilization is fast with the flow of air. Thus, the introduction of flowing gas, raising the temperature, facilitates the evaporation of free water from the organic precursors.
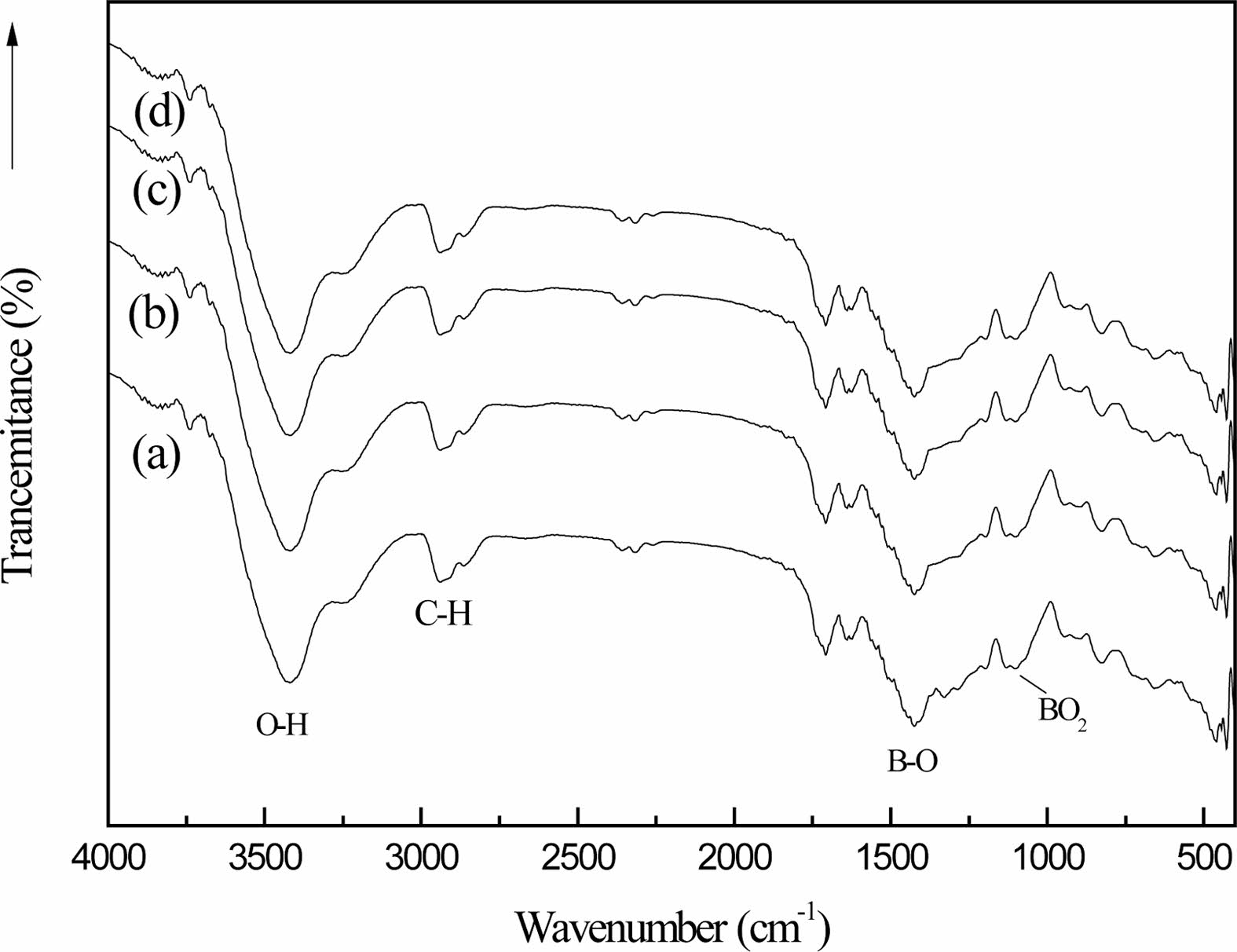
Fig. 8 shows the infrared spectra of the organic precursors in different cracking atmospheres (static air (a), air flow (b), nitrogen flow (c) and argon flow (d)) after 2h heat treatment at 250 ℃ and then 2h heat treatment at 350 ℃. Fig. 8 and Fig. 1(c) are compared shows the stretching vibration peak of B-O-C bond at 1050 cm-1 and the stretching vibration peak of B-OH at 1189 cm-1 are obviously weakened. In addition, the bending vibration peak of O-H at 3196 cm-1 also becomes very weak, and the bending vibration peak of BO2 appears at 1120 cm-1, which is the characteristic absorption peak of boron oxide [29]. These characteristics indicate that the organic precursors have undergone chemical changes after two steps of heat treatment.
The stretching vibration peak of C-H bond appears at 2913 cm-1, which proves that the chemical bond of the organic precursor is broken. The vibration peak strength of BO2 bond in curve (b) is greater than that in curves (a), (c) and (d), and the vibration peak strength of BO2 bond in curve (a) is the weakest. Through infrared spectrum analysis of samples of the same mass, it can be quantitatively analyzed that the reaction rate of organic precursors in the atmosphere with airflow is faster than that in the still air [6].
Preliminary carbonization of organic precursors has occurred. According to TG curve analysis, increasing reaction temperature is beneficial to carbonization of organic precursors. The pyrolysis products were treated at 500 ℃, 550 ℃, 600 ℃, 650 ℃, 700 ℃, 750 ℃ and 800 ℃ for 2 h, respectively, and analyzed by Fourier transform infrared spectroscopy.
Fig. 9 shows the infrared spectra of pyrolysis products after initial carbonization of organic precursors in different pyrolysis atmospheres (static air (a), air flow (b), nitrogen flow (c) and argon flow (d)) and 2 h constant temperature heat treatment at pyrolysis temperature of (500-800 ℃). The bending vibration peak of the BO2 bond at 1120 cm-1 and the stretching vibration peak of the B-O bond at 1454 cm-1 of the pyrolysis products gradually increase with the increase of temperature. The stretching vibration peak of C-H bond at 2913 cm-1 disappeared, and the vibration absorption peak of O-H bond at 3000-3500 cm-1 shifted with the change of temperature. This is because there is still trace amount of mono hydrate boric acid in the pyrolysis products [17, 28].
Compared (a), (b), (c) and (d) in Fig. 8, it can be seen that when the cracking temperature reaches 550 ℃, the stretching vibration peak of B-O-C bond at the wave-number of 1050 cm-1 and the stretching vibration peak of B-OH bond at the wave-number of 1189 cm-1 still exist in (a), (c) and (d), but the absorption peak intensity is very weak. However, the two stretching vibration peaks in (b) have disappeared, indicating that the organic precursors in (b) have cracked completely at this time. When the cracking temperature reaches 650 ℃, B-O-C bond and B-OH bond with weak absorption peak intensity still exist in (a), while in (C) and (d), these two stretching vibration peaks have disappeared, indicating that the organic precursors in (C) and (d) have been completely cracked at this time. When the pyrolysis temperature reached 750 ℃, the stretching vibration peaks of B-O-C and B-OH bonds could not be found in (a), (b), (c) and (d), indicating that the organic precursors had been completely cracked at this temperature. When the cracking atmosphere is air flow, the organic precursors are completely cracked at 550 ℃; when the temperature reaches 650 ℃, the organic precursors in nitrogen flow and argon flow are completely cracked; and when the cracking temperature reaches 750 ℃, the organic precursors in static air are completely cracked. This fully shows that during the cracking process, The flowing gas is helpful to the improvement of cracking efficiency, and the cracking efficiency in air flow is the highest. The disappearance of the stretching vibration peaks of B-O-C bond and B-OH bond strongly confirms that the organic precursors have undergone cracking changes.
After cracking in different atmosphere, the phase structure of the final cracking products is basically the same. At the same time, in the infrared spectrum of pyrolysis products, there is no absorption peak of B-C bond stretching vibration, indicating that the organic precursors did not generate B4C after three steps of heat treatment reaction.
Microstructure analysis of pyrolysis products
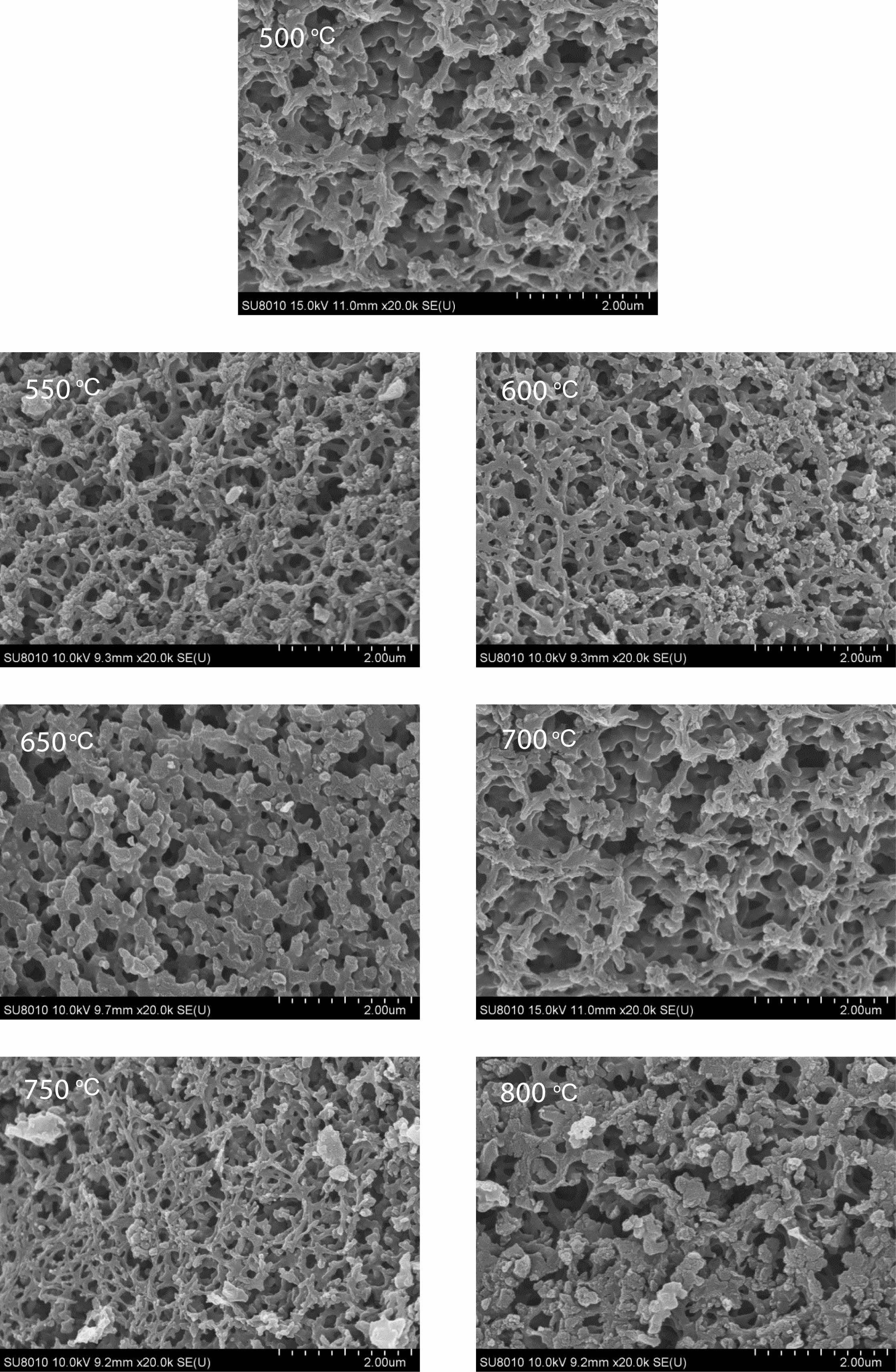
Morphology analysis of pyrolysis products of free carbon after removal of boron oxide by hot water washing. Fig. 10 shows the SEM spectrum of the residue after hot water washing and boron removal of the pyrolysis products prepared at different cracking temperatures (500-800 ℃) in still air under cracking atmosphere. After the pyrolysis products are washed in hot water and boron oxide is removed, the remaining products show a reticular structure. With the increase of temperature, the reticular structure becomes more and more dense and the pores become more and more regular, and the pore size is all less than 1 um. Fig. 11 shows organic precursor pyrolysis products in the air flow through the hot water washing the rest of the carbon residue still rendering of three-dimensional mesh structure, but with the increase of temperature, the reticular structure first become dense, and then become loose, after the first hole size is smaller, especially 550 ℃ heat treatment have been the product of pyrolysis product after hot water washing, get the most dense mesh structure, and the The diameter of the hole is the smallest, reaching below 0.1 um. Fig. 12 shows organic precursor pyrolysis product in the nitrogen flow with the increase of temperature, carbon residue mesh structure first become dense, become loose, after 650 ℃ heat treatment have been the product of pyrolysis product after hot water washing, carbon network structure is most dense, aperture below minimum can be up to 0.1 um, but each image in the aperture size is highly structured, At 800 ℃, the maximum pore size of carbon network can reach 2 um. Fig. 13 shows organic precursor in the argon gas flow split product residue after hot water washing except boron, with the increase of temperature, have been maintaining a state of dense mesh structure, however, this kind of mesh structure of aperture with the decrease of temperature rises bigger before, 650 ℃ heat treatment have been the pyrolysis product residue after hot water washing, reached the maximum aperture, Is about 1.5 um, and the pore size of the carbon network is below 0.1 um at 500 ℃.
The comparison of Fig. 10-13 shows that the carbon net structure of cracking products in air flow is the most dense and the pore size is the most uniform. The carbon net structure of cracking products in static air is also very dense, but the pore size of carbon net is much larger than that of cracking products in air flow. By washing the powder of pyrolysis product repeatedly in hot water at 80 ℃, the composition of B2O3 can be removed, and the amorphous free carbon with special carbon network structure with micron spacing can be obtained. This unique structure is similar to macroporous materials, in esterified derivatives, through phase separation to form a three-dimensional double continuous structure
The double continuous network structure of residual carbon residue makes the contact sites between free carbon and B2O3 increase obviously. The structure increases the reactivity and nucleation center number of B4C, which is beneficial to decrease the synthesis temperature of B4C. This dual continuous network structure is generated in the condensed H3BO3-C3H8O3 and remains in the pyrolysis product powder, which means that the pyrolysis process is important. The morphology and particle size of the prepared boron carbide are affected by the dual continuous network structure with different densities.
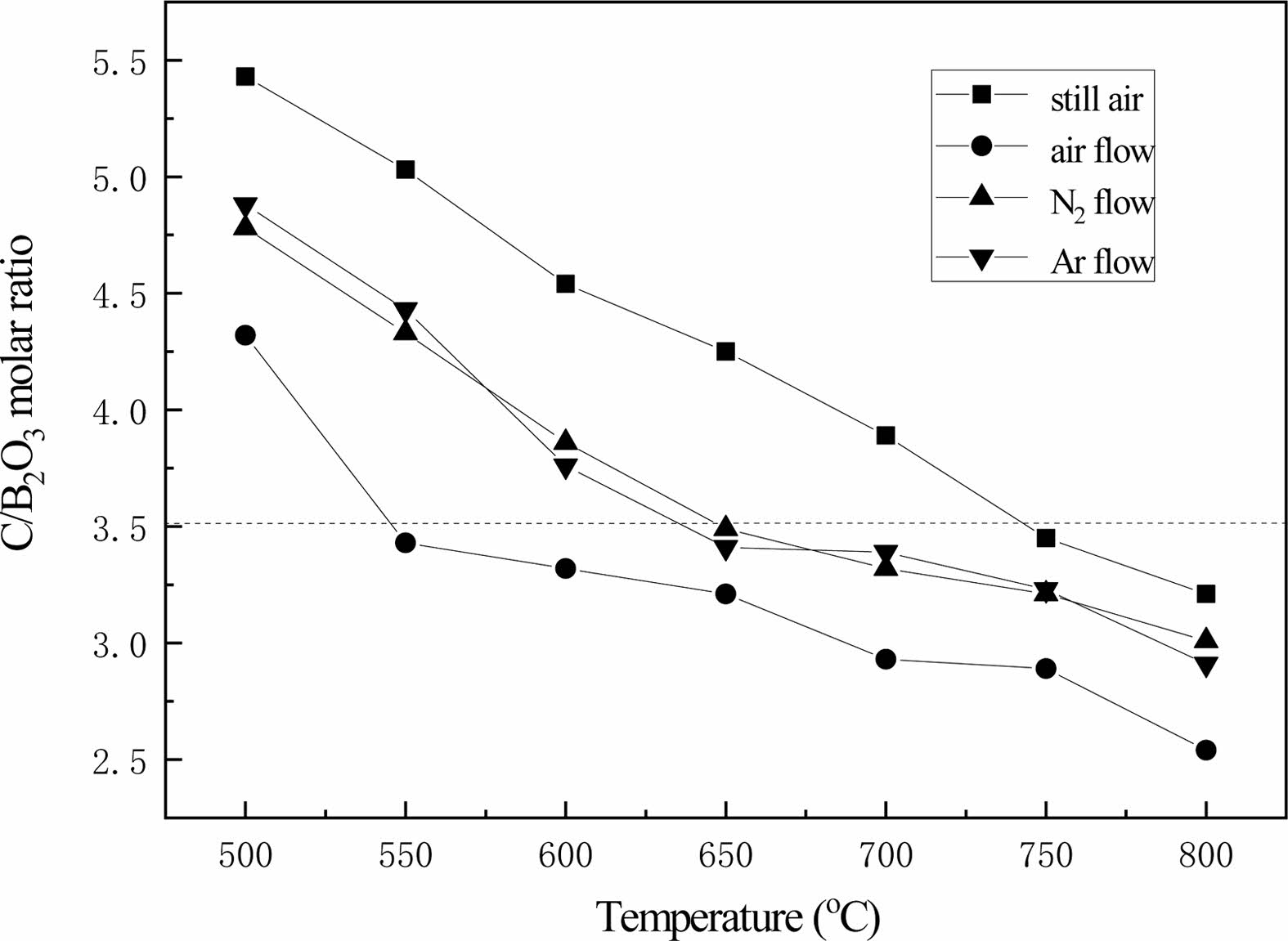
Fig. 14 shows the changing curve of the C/B2O3 molar ratio of pyrolysis products. With the increase of pyrolysis temperature, the C/B2O3 molar ratio of pyrolysis products decreases significantly. This is due to the high temperature, the reaction rate of carbide and oxygen in the air is accelerated, and the oxygen consumption of carbon increases in the same time. In nitrogen flow and argon flow, free carbon is blown out along with the air flow, so that the equilibrium of chemical reaction is carried out in the forward direction. Through quantitative analysis, it is found that the molar ratio of C/B2O3 of pyrolysis products in the static air is obviously higher than that of pyrolysis products in the air through any temperature comparison. This indicates that air flow can improve the cracking efficiency of organic precursors.
According to Eq. (1), if the pyrolysis products are reduced to produce boron carbide at high temperature, the pyrolysis products with a C/B2O3 molar ratio slightly less than 3.5 should be selected. The pyrolysis temperature of the organic precursors is 750 ℃, and the molar ratio of C/B2O3 is 3.45. The molar ratios of C/B2O3 were 3.49 and 3.41, respectively, when the pyrolysis temperature was 650 ℃ in nitrogen and argon. When cracking in air flow, the cracking temperature is 550 ℃, and the molar ratio of C/B2O3 obtained is 3.43. The molar ratio of C/ B2O3 obtained is close to but less than 3.5.
Therefore, these four pyrolysis products were selected as raw materials for the preparation of B4C by carbothermal reduction reaction.
Crystalline phase analysis of B4C
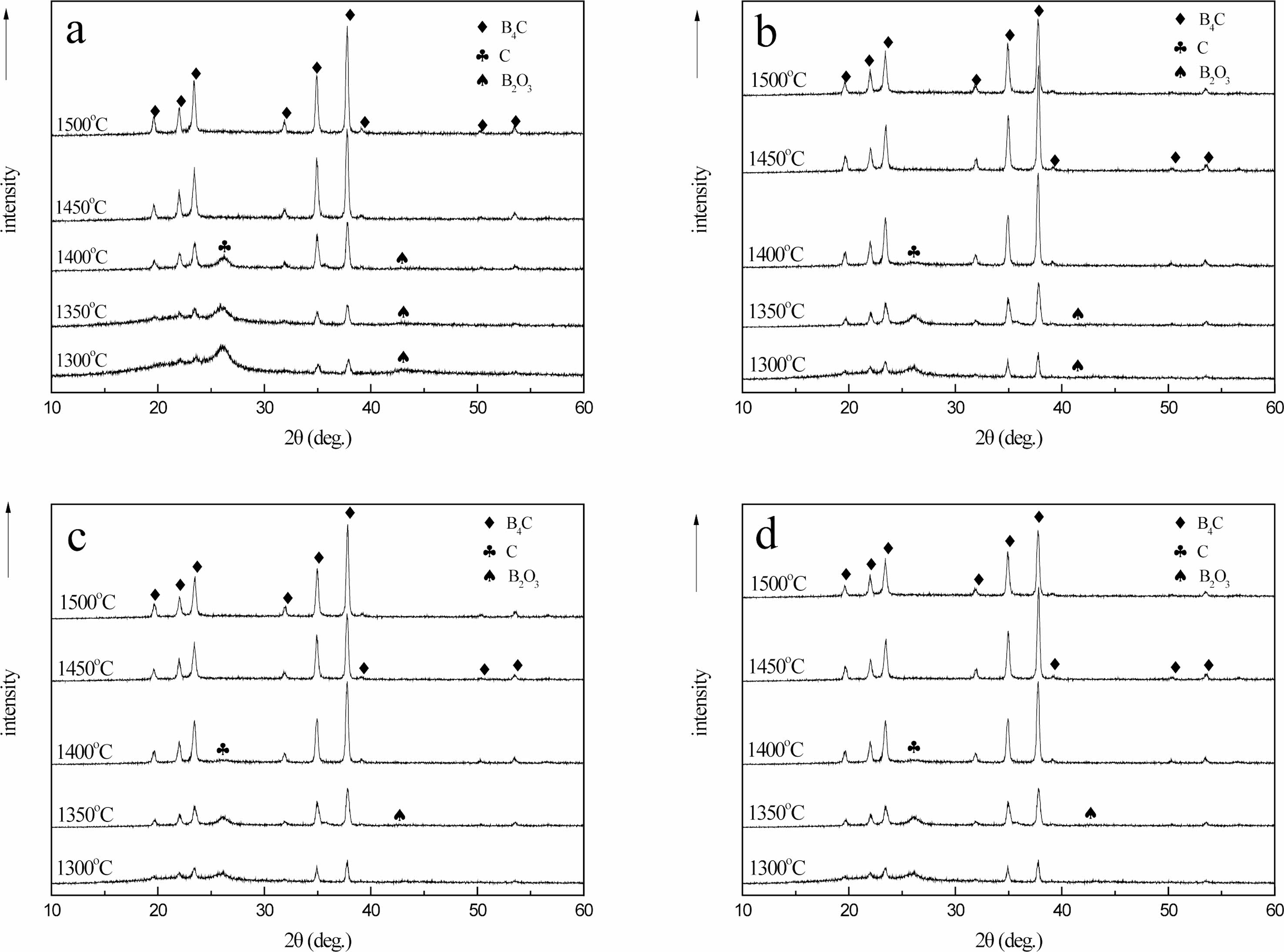
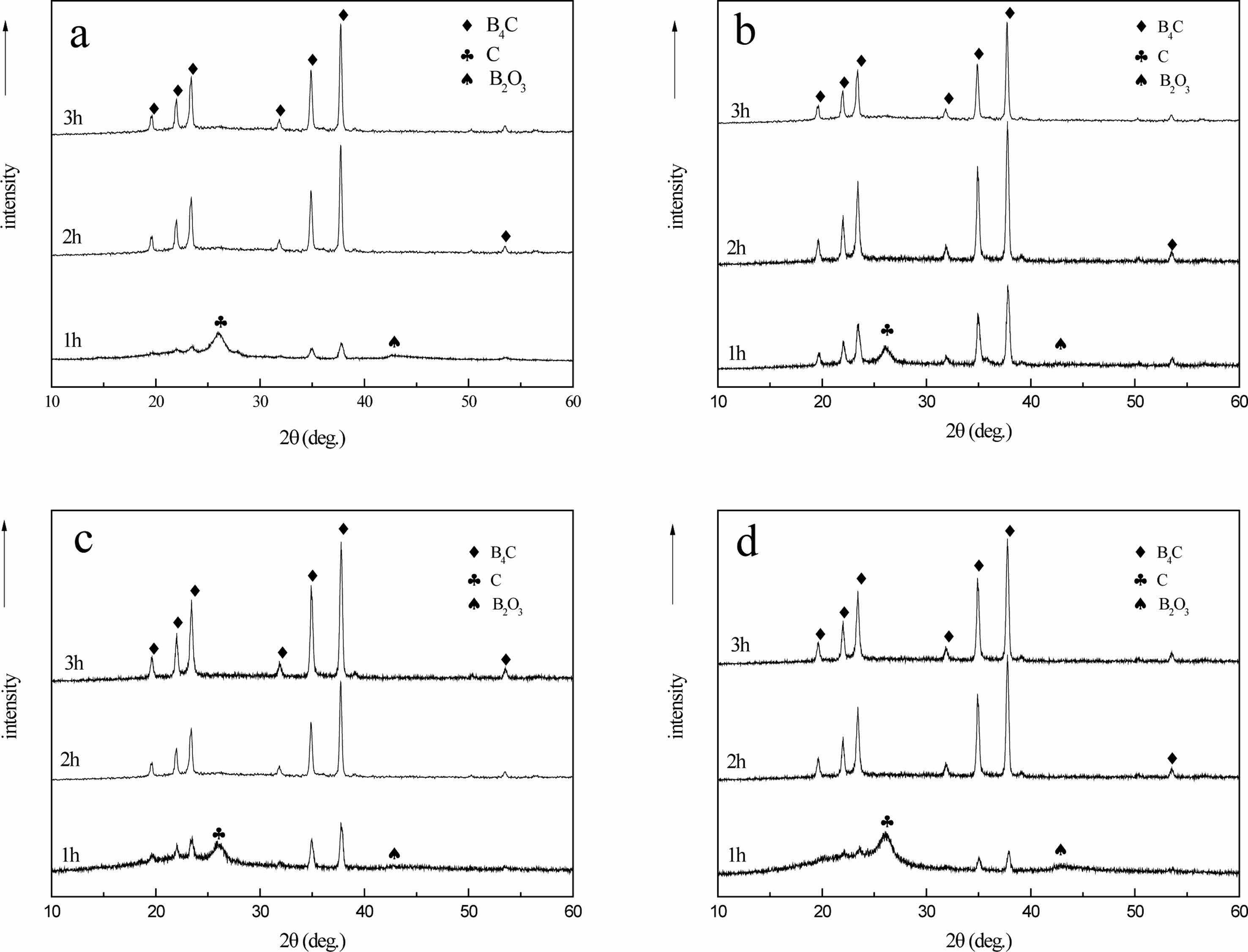
Fig. 15 shows that when the carbothermal reduction temperature is 1300 ℃, the characteristic diffraction peaks of B4C appear at 2θ=34.8o and 37.7o. With the increase of reaction temperature, the diffraction peaks of B4C crystal phase gradually increase. This is because with the increase of temperature, the crystallization driving force of B4C also increases, leading to the gradual enhancement of diffraction peak intensity. The crystallinity was improved. At 1300-1400 ℃, there are characteristic diffraction peaks of free carbon and boron oxide at 26.6o and 41.6o of 2θ, and the raw materials are not completely reacted.
When the temperature rises to 1450-1500 ℃, the diffraction peaks of 19.6°, 21.9°, 23.3°, 31.8°, 34.9°, 37.7°, 39.1°, 50.1° and 53.4° of 2θ are characteristic diffraction peaks of crystal B4C. Corresponding to (101), (003), (012), (110), (104), (021), (113), (211) and (205) crystal faces, respectively [24].
The diffraction data and the peak value of the standard card (JCPDSNo. 35-0798) of crystal B4C have a high matching degree, indicating that the sample prepared under this condition is B4C crystal. It is further confirmed that the B4C belongs to the hexagonal system and has a rhombic crystal structure. The characteristic peaks of free carbon and boron oxide have disappeared. At this time, pure B4C is prepared. It can also be seen that the peak shape of each diffraction peak is relatively sharp, and the intensity of the diffraction peak is very strong, indicating that the crystallinity of the product is high and the crystal form is relatively complete.
When the carbothermal reduction temperature is 1400 ℃, the characteristic diffraction peak of B4C appears at 50.1° for 2θ in (b), (c) and (d), while the existence of diffraction peak is not observed at the same position in (a), indicating that the crystal growth of B4C in (b), (c) and (d) is more complete than that in (a). When 2θ is 37.7°, only the characteristic diffraction peak of B4C appears in (b), indicating that the crystal growth of B4C is the best in (b) at this time. When the carbothermal reduction temperature rises to 1450 ℃, the B4C crystals in (a), (b), (c) and (d) grow completely and have a complete crystal form. Compared with the diffraction peak intensity of 34.9° 2θ, the peak strength of (a) is 7689.1, (b) is 9533.6, (c) is 8534.2, and (d) is 8722.4. During the XRD test, the mass of samples is 0.1 g, and the qualitative analysis can be carried out. In (b), the crystallinity of B4C is the best, followed by (d), (c), while in (a), the crystallinity of B4C is slightly worse.
The possible reason is (a) for the air flow in the pyrolysis products as a raw material, the density of carbon and uniform carbon mesh aperture, is beneficial to the growth of B4C, make the boron carbide forming fast, and crystalline is best, with nitrogen and argon gas flows in pyrolysis products as raw material, its carbon mesh aperture uneven, density is a bit poor, make the boron carbide crystal properties affected by certain, When cracking products in static air are used as raw materials, due to the large pore size of the carbon network, the combination of boron oxide and free carbon is more difficult, which makes the forming of B4C difficult and the crystallization deviation.
The pyrolysis products obtained in different pyrolysis atmosphere will have a certain influence on the molding and crystallizability of B4C, and the pyrolysis products in air flow can be used as raw materials to prepare boron carbide, which can produce B4C with fast molding speed and high crystallizability.
Fig. 16 shows that when the time of carbothermal reduction reaction is 1 h, two characteristic diffraction peaks of B4C with weak peaks appear at 34.8° and 37.7° of 2q, and there are diffraction peaks of free carbon and boron oxide with 26.6° and 41.6° of 2q, respectively. With the prolongation of reaction time, the characteristic diffraction peak strength of B4C gradually increases, and the characteristic diffraction peak strength of free carbon and boron oxide decreases. Prolonging reaction time can promote the further reaction of C and B2O3. When the time of carbothermal reduction is 2 h, it can be observed from the XRD spectrum that the peak of carbon and boron oxide has disappeared, which proves that the carbothermal reduction reaction is complete at this time.
The characteristic diffraction peaks of crystal B4C with complete peaks of 19.6°, 21.9°, 23.3°, 31.8°, 34.9°, 37.7° and 53.4° at 2θ. After further extending the reaction time to 3 h, the peak shape and position of characteristic diffraction peak of B4C did not change significantly.
Product crystallinity analysis
The theoretical expression (Eq. 2) of the relationship between B4C peak intensity ratio and crystallinity obtained by Naoki Tahara et al. The larger the I, the better the crystallinity of B4C, the smaller the I, the worse the crystallinity of B4C.
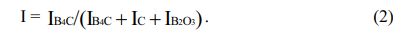
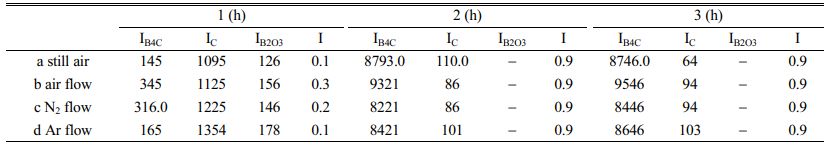
Table 1 shows that when the carbothermal reduction time is 1 h, the I values of a-d are 0.1, 0.3, 0.2 and 0.1, respectively, indicating that the crystallinity of B4C in the product is very poor at this time. However, when the reaction time is extended to 2 h, the I values of a-d reach 0.9, indicating that the crystallinity of B4C at this time is already very high.
When the reaction time was prolonged, the peak intensity of B4C and the value of I did not change significantly. From the perspective of industry, in order to save energy and reduce energy consumption, 2 h of carbothermal reduction time was selected as the appropriate time to prepare B4C.
According to the above study on the carbothermal reduction temperature and reaction time in the preparation process of boron carbide, under the conditions of carbothermal reduction temperature of 1450 ℃ and reaction time of 2 h, B4C powder with good particle dispersion, excellent morphology and high purity can be prepared
Morphology analysis of the product
Fig. 17 shows the final B4C from different prolysized atmosphere (a) still air, (b) air flow, (c) N2 flow, (d) Ar flow. When the pyrolysis products obtained in static air are used as raw materials, the morphology is polyangular type and the particle size is about 5.7 um B4C powder. When the pyrolysis products obtained in air flow are used as raw materials, the morphology is smooth and granular B4C powder with a particle size of about 1.5 um. When the pyrolysis products obtained in nitrogen flow are used as raw materials, the morphology is granular, flake and rod-like particles, and the size of B4C powder is about 14.0 um. When the pyrolysis products obtained in argon flow are used as raw materials, the B4C powder with hexagonal shape and particle size of about 23.3 um is obtained.
This formed a corresponding relationship with the carbon network structure of the pyrolysis products prepared, and the carbon residue analysis of the material as the raw material of carbothermal reduction reaction of boron carbide in Fig. 10-13 could be compared. The pyrolysis products consist of boron oxide and amorphous free carbon. When the pyrolysis atmosphere is still air and the pyrolysis temperature is 750 ℃, the structure of the carbon net is the loosest, and the pore size of the carbon net is relatively uniform around 1 um. When the pyrolysis atmosphere is flowing air, the carbon net structure is the densest at the pyrolysis temperature of 550 ℃, and the pore size of the carbon net is uniformly about 0.1 um. When the pyrolysis atmosphere is flowing nitrogen and flowing argon, 650 ℃ is the suitable pyrolysis temperature. The density of the carbon network structure is between the first two, and the pore size of the carbon network is different. This fully shows that the diameter of carbon net is directly related to the growth state of boron carbide particle size.
Particle size analysis of B4C
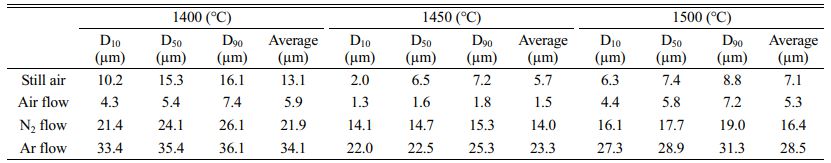
Table 2 shows that when the pyrolysis products obtained in different cracking atmospheres (static air, air flow, nitrogen flow and argon flow) are used as raw materials for the preparation of B4C, the size of the particles decreases first and then increases when the carbothermal reduction temperature increases from 1400 ℃ to 1500 ℃.
When the temperature is 1400 ℃, the B4C particles are not completely separated from the carbon network, and there are a lot of free carbon and boron oxide in the product. When the temperature rises to 1500 ℃, some B4C particles agglomerate, making the particle size significantly increase, which is consistent with the FE-SEM map. B4C particles with average particle size of 5.7 um, 1.5 um, 14.0 um and 23.3 um can be prepared by using pyrolysis products in different atmospheres (static air, air flow, nitrogen flow and argon flow) when the carbothermal reduction temperature is 1450 ℃. The size of B4C particles obtained is the smallest when the pyrolysis products in the air flow are used as raw materials for carbothermal reduction, while the size of B4C particles obtained is the largest when the pyrolysis products in the argon flow are used as raw materials for carbothermal reduction.
This is basically consistent with the particle size observed in FE-SEM above, which fully proves that the particle size of B4C is closely related to the carbon network structure in the pyrolysis products of the used raw materials. B4C particles with different particle sizes can be prepared from the pyrolysis products in different pyrolysis atmospheres through the same carbothermal reduction reaction.
Purity analysis of B4C
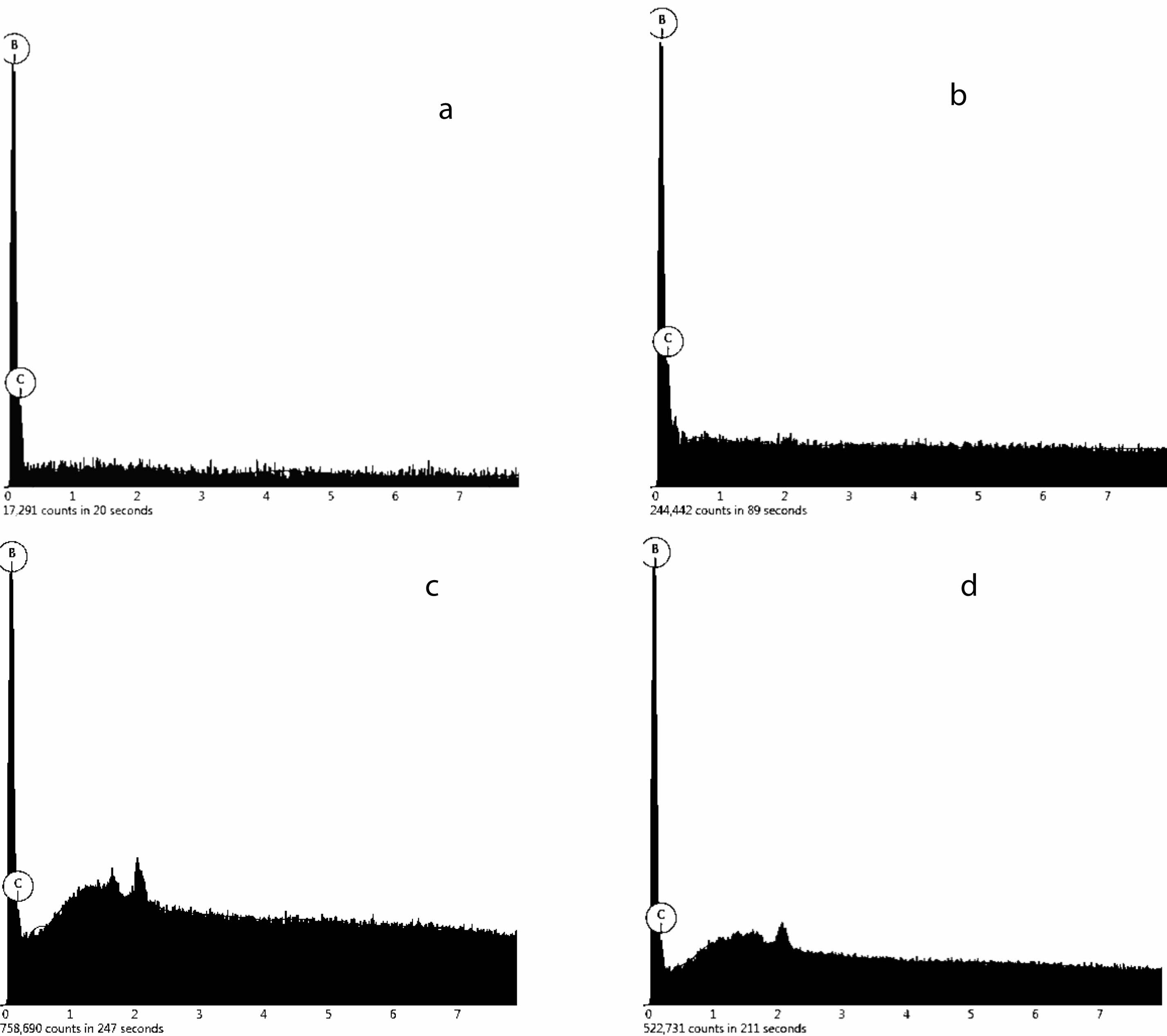
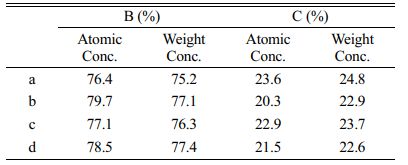
Fig. 18 and Table 3 shows weight concentration of element in samples. The pyrolysis products obtained from static air (A), air flow (B), nitrogen flow (C) and argon flow (D) are used as raw materials, and the products are prepared by carbothermal reduction. The mole fraction of B was 76.4%, 79.7%, 77.1%, 78.5%, respectively. The molar fractions of C were 23.6%, 20.3%, 22.9% and 21.5%, respectively. The molar ratio of B to C is about 4:1.23, 4:1.02, 4:1.18, 4:1.09; It indicates that the final product contains a small amount of free carbon, and the mass content of B4C in the final product is calculated to be 95.9%, 98.9%, 96.4% and 97.1%. The B2O3 in the pyrolysis products of air flow is embedded in the amorphous carbon dense network structure, which reduces the volatilization loss of B2O3 and makes the reaction degree more complete. However, in still air, the network aperture of amorphous carbon is relatively large, which makes the B2O3 embedded in it easier to volatilization, so there is more free carbon left. However, the carbon network structure of cracking products obtained in nitrogen and argon flows is neither as dense as that of cracking products in air flow, nor as loose as that of carbon network structure in static air, so the content of free carbon is between the both.
|
Fig. 1 FT-IR spectra of glycerol (a), boric acid (b) products (c). |
|
Fig. 2 TGA curves of boric acid (a), glycerol (b) and condensed products (c). |
|
Fig. 3 Reaction mechanism of boric acid with glycerol. |
|
Fig. 4 Effect of glycerol to boric acid molar ratio on the conversion of boric acid. |
|
Fig. 5 Effect of the reaction temperature on the conversion of boric acid. |
|
Fig. 6 Effect of the reaction time on the conversion of boric acid |
|
Fig. 7 FT-IR spectra of polymeric precursors heat treatment at 250 ℃ for 2 h in still air (a), air flow (b), nitrogen flow (c) and argon flow (d). |
|
Fig. 8 FT-IR spectra of polymeric precursors heat treatment at 250 ℃ for 2 h and 350 ℃ for 2 h in still air (a), air flow (b), nitrogen flow (c) and argon flow (d). |
|
Fig. 9 FT-IR spectra of polymeric precursors heat treatment at different temperatura in still air (a), air flow (b), nitrogen flow (c) and argon flow (d). |

|
Fig. 10 SEM image of pyrolysis product after hot water washing heat treated at different temperature in still air. |
|
Fig. 11 SEM image of pyrolysis product after hot water washing heat treated at different temperature in air flow |

|
Fig. 12 SEM of pyrolysis product after hot water washing heat treated at different temperature in argon flow. |

|
Fig. 13 SEM of pyrolysis product after hot water washing heat treated at different temperature in nitrogen flow |
|
Fig. 14 Change in C/B2O3 molar ratio of precursor powders |
|
Fig. 15 XRD patterns of B4C prepared at different carbothermal reduction temperatures by pyrolysis products in still air (a), air flow (b), nitrogen flow (c) and argon flow (d). |
|
Fig. 16 XRD patterns of B4C prepared at different carbothermal reduction time by pyrolysis products in still air (a), air flow (b), nitrogen flow (c) and argon flow (d) |

|
Fig. 17 FE-SEM image of B4C the prolysis products from different prolysized atmosphere (a) still air,(b) air flow, (c) N2 flow, (d) Ar flow. |
|
Fig. 18 EDS patterns of B4C prepared products heated treatment at 1450 ℃ by pyrolysis products in still air (a), air flow (b), nitrogen flow (c) and argon flow (d) |
|
Table 1 Comparative tables of crystallinity of different pyrolysis products and carbothermal reduction at different reaction time. |
In this paper, the influence of pyrolysis atmosphere on the morphology and particle size of boron carbide was studied. In different pyrolysis atmospheres, the pore size of the three-dimensional carbon network structure obtained is different, which leads to the different number of boride attachment sites, and affects the size and morphology of boron carbide. The carbonization process of borate glyceride can be accelerated by air flow is the most obvious, which makes the carbon network structure extremely dense. Therefore, the narrowest particle size of 1.5um boron carbide can be obtained. There is no airflow in the still air, and the pyrolysis products can not be fully taken away, which prolongs the carbonization process and makes the carbon network loose. Therefore, the particle size of the product boron carbide is quite high. According to the influence of different airflow, different size of boron carbide particles can be customized according to the requirements. And affected by nitrogen flow, boron carbide can form sheet structure. In the pyrolysis products, the carbon network structure is dense, and the carbon network with small pore size will obtain the B4C powder with uniform particle morphology and small particle size.
The authors would like to acknowledge financial support from Key Laboratory of Liaoning Province for Polymer Catalytic Synthesis, Liaoning Provincial Engineering Laboratory for Advanced Polymeric Materials and High-tech Research and Development Project of Liaoning Provincial Industrial Special Resources Protection Office (Liao Cai Qi [2013]736).
- 1. K.H. Kim, K.B. Shim, J.H. Chae, J.S. Park, and J.P. Ahn, J Ceram Process Res. 10[6] (2009) 716-720.
- 2. P.G. Krishnan, B.S. Babu, S. Madhu, S.J. Gowrishankar, C. Bibin, S. Saran, S.S. Ram, A.R.S. Hari, and S. Vidyasagar, J Ceram Process Res. 22[5] (2021) 483-489.
-

- 3. P. Svec, Z. Gabrisova, and A. Brusilova, J Ceram Process Res. 20[1] (2019) 113-120.
- 4. K.H. Kim, J.H. Chae, J.S. Park, D.K. Kim, K.B. Shim, and B.H. Lee, J Ceram Process Res. 8[4] (2007) 238-242.
- 5. A.K. Suri, C. Subramanian, J.K. Sonber, and T.S.R.C. Murthy, Int. Mater. Rev. 55[1] (2010) 4-40.
-
- 6. Werheit, and Helmut, Solid State Sci. 60 (2016) 45-54.
-
- 7. C. Cheng, K.M. Reddy, A. Hirata, T. Fujita, and M. Chen, J. Eur. Ceram. Soc. 37[15] (2017) 4514-4523.
-
- 8. K.Y. Xie, V. Domnich, L. Farbaniec, B. Chen, K. Kuwelkar, L.N. Ma, G.W. McCauley, R.A. Haber, K.T. Ramesh, M.W. Chen, and K.J. Hemker, Acta Mater. 136 (2017) 202-214.
-
- 9. J.U. Mohamed, P.K. Palaniappan, P.B. Maran, and R. Pandiyarajan, J Ceram Process Res. 22[3] (2021) 306-316.
-
- 10. F. Farzaneh, F. Golestanifard, M.S. Sheikhaleslami, and A.A. Nourbakhsh, Ceram. Int. 41[10] (2015) 13658-13662.
-
- 11. A. Sudoh, H. Konno, H. Habazaki, and H. Kiyono, Carbon. 45[6] (2010) 1373-1373.
-
- 12. A. Sinha, T. Mahata, and B.P. Sharma, J. Nucl. Mater. 301[2-3] (2002) 165-169.
-
- 13. J.L. Parsons and M.E. Milberg, J. Am. Ceram. Soc. 43[6] (2006) 326-330.
-
- 14. A.M. Hadian and J.A. Bigdeloo, J. Mater. Eng. Perform. 17[1] (2008) 44-49.
-
- 15. N. Tahara, M. Kakiage, I. Yanase, and H. Kobayashi, J. Alloys Compd. 573[1] (2013) 58-64.
-
- 16. A. Najafi, F. Golestani-Fard, H.R. Rezaie, and N. Ehsani, J. Alloys Compd. 509[37] (2011) 9164-9170.
-
- 17. S. Mondal and A.K. Banthia, J. Eur. Ceram. Soc. 25[2-3] (2005) 287-291.
-
- 18. I. Yanase, R. Ogawara, and H. Kobayashi, Mater. Lett. 63[1] (2009) 91-93.
-
- 19. M. Kakiage, N. Tahara, I. Yanase, and H. Kobayashi, Mater. Lett. 65[12] (2011) 1839-1841.
-
- 20. M. Kakiage, N. Tahara, S. Yanagidani, I. Yanase, and H. Kobayashi, J. Ceram. Soc. Jpn. 119[1390] (2011) 422-425.
-
- 21. M. Kakiage, Y. Tominaga, I. Yanase, and H. Kobayashi, Powder Technol. 221 (2012) 257-263.
-
- 22. B. Wade, N. Venkatasubramanian, P. Desai, A.S. Abhiraman, and L.T. Gelbaum, J. Sol-gel. Sci. Techn. 5[1] (1995) 15-25.
-
- 23. N. Shawgi, S.X. Li, S. Wang, Y. Li, and R. Ramzi, Ceram. Int. 44[8] (2018) 9887-9892.
-
- 24. A. Najafi, F. Golestani-Fard, H.R. Rezaie, and N. Ehsani, Ceram. Int. 38[5] (2012) 3583-3589.
-
- 25. N. Shawgi, S.X. Li, S. Wang, Y. Li, and R. Ramzi, Ceram. Int. 44[1] (2018) 774-778.
-
- 26. M.M. Balakrishnarajan, P.D. Pancharatna, and R. Hoffmann, New J. Chem. 31[4] (2007) 473-485.
-
- 27. A.K. Khanra, B. Mater. Sci. 30[2] (2007) 93-96.
-
- 28. C. Sun, X.G. Lu, Y.B. Chen, L.L. Zuo, and Y.K. Li, J Ceram Process Res. 22[3] (2021) 340-344.
-
- 29. H.K. Wei, Y.J. Zhang, and X.Y. Deng, J Ceram Process Res. 12[3] (2011) 599-601.
- 30. A.M. Hadian, and J.A. Bigdeloo, J. Mater. Eng. Perform. 17[1] (2008) 44-49.
-
 This Article
This Article
-
2022; 23(5): 595-610
Published on Oct 31, 2022
- 10.36410/jcpr.2022.23.5.595
- Received on Feb 23, 2022
- Revised on Apr 16, 2022
- Accepted on May 7, 2022
Services
- Abstract
introduction
experimental
results and discussion
conclusion
- Acknowledgements
- References
- Full Text PDF
Shared
Correspondence to
- Li Yang, Li Sanxi
-
School of Environmental and Chemical Engineering, Shenyang University of Technology, Liaoning, 110870, China
Tel : +86 18704009103 - E-mail: lisx@sut.edu.cn
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr